
Introduction to Computational Structural Biology Virtual Workshop
Here, we present our new workshop titled “Introduction to Computational Structural Biology Virtual Workshop” which will be held on 18-19 December 2021. Our workshop will be conducted online (by Zoom) due to Covid19. So, we encourage researchers who are interested in structural biology, protein structure, protein-protein docking, protein-ligand docking and molecular modeling simulation to register for this virtual workshop. The workshop will be conducted by two main sessions. The first day of the workshop will start with theory sections and cover the experimental techniques in protein structure determination, and molecular docking. After the theory section, we will continue with the hands-on part which will be based on an explanation of protein structure files, molecular visualization tools, and implementation of molecular docking by using HADDOCK, SWISS and PRISM. The second day will present the theory section of molecular modeling & simulation with the hands-on section which will be based on the practice of NAMD simulation and analysis of MD simulations data. The workshop will be ended with virtual party and networking session.
Instructors of Theory Sections:

Asst. Prof. Burak V. Kabasakal
Principal Investigator at Turkish Accelerator and Radiation Laboratory (TARLA), Ankara University
Theory: Experimental Methods for Identification of Protein Structure
Experimental techniques used in protein structure determination will be presented. High-resolution structure determination methods; X-ray Crystallography, Cryogenic electron microscopy (Cryo-EM), Nuclear magnetic resonance spectroscopy (NMR) and the supplementary low-resolution method Small Angle X-ray Scattering (SAXS) will be explained.

Asst. Prof. Saliha Ece Acuner
Bioengineering Dept. at Istanbul Medeniyet University
Theory: Molecular and Macromolecular Docking
Proteins are essential for the cell by performing various functions and they seldom act alone. Interactions of proteins with other molecules can be determined experimentally but simple large scale methods can only spot whether a pair is interacting or not. However, structural details of the complex are crucial for effective drug design. The structure of the complex can be determined experimentally but it takes a long time and sometimes impossible if the complex is transiently interacting. Molecular modeling, specifically docking, aims to address this problem by computationally predicting protein-small molecule, protein-protein or protein-nucleic acid complex structures.

Prof. Dr. E. Demet Akten Akdoğan
Bioinformatics and Genetics Dept. at Kadir Has University
Theory: Giant Workers in Cells: Proteins and Their Specific Dynamic Structures
Proteins are long polymeric chains of different types that consist of thousands of atoms and have different functions in cells. While experimental methods can provide only a snapshot of these giant molecules, a Molecular Dynamics (MD) simulation using an atomistic model can record the motion of each atom over time in a computer setting. By introducing a physics-based interaction model, the analysis of a simulation performed on a GPCR (G-Protein Coupled Receptor) protein family will be presented. In the second part of the talk, the allosteric (remote-controlled) feature, which is an important part of the protein’s dynamic structure will be introduced. How allosteric information can be used in drug development studies that will only target bacteria or parasites instead of the host organism will be presented for glycolytic enzymes.
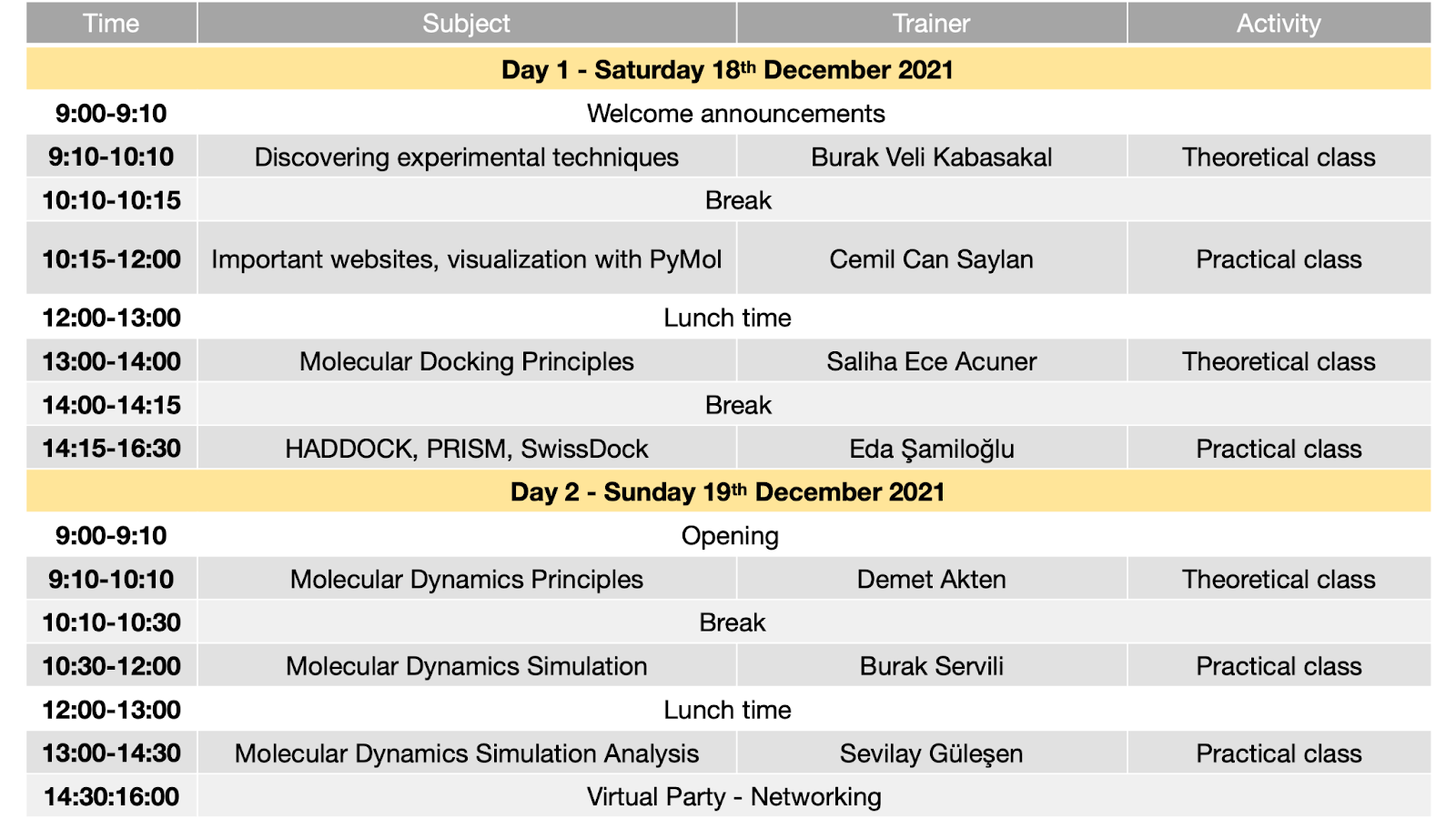
Program
Registration