
What are the omics sciences?
Omics sciences are targeting quantification of whole biomolecules such as RNA and proteins at organism, tissue, or a single-cell level. Omics sciences are separated into several branches such as genomics, transcriptomics, and proteomics1.
What is transcriptomics?
Transcriptomics is one of the omics sciences dissecting the organism’s transcriptome which is the sum of all of its RNA molecules2,3.
What is RNA sequencing?
RNA sequencing (RNA-seq) is a technique providing quantification of all RNAs in bulk tissues or each cell. The transcript amounts of each gene across samples are calculated by using this technique. It is utilizing next-generation sequencing (NGS) platforms deciphering the sequencing of biomolecules such as DNA and RNA4,5.
What are the kinds of RNA-seq?
Bulk tissue RNA-seq
The whole transcriptome of target bulk tissues is sequenced to make transcriptomics analyses. Here, target bulk tissue can contain various cell types, and therefore, the whole transcriptome is mixed with RNAs of those cells. This approach is the most common usage of RNA-seq and is performed for some aims such as elucidating of diseases7.
Single-cell RNA-seq
In contrast to bulk tissue RNA-seq, single-cell RNA-seq (scRNA-seq) is performed in individual cells. The whole transcriptome of each cell in a tissue is sequenced to make transcriptomics analysis. The scRNA-seq has revealed that the transcriptome of each cell in a tissue is different from each other and individual cells can be separated into specific clusters according to its transcriptomic signature. The scRNA-seq has helped the discovery of some cells such as ionocyte cells, which could be relevant to the pathology of cystic fibrosis7,8.
Spatial RNA-seq
The relationship between cells and their relative locations within a tissue sample can be critical to understanding disease pathology. Spatial transcriptomics is a technology that allows the measurement of all the gene activity in a tissue sample and map where the activity is occurring. This technique is utilized in the understanding of biological processes and disease. Spatial RNA-seq can be performed at intact tissue sections as well as a single-cell level. The general aim of this technique is a combination of gene expression and morphological information and providing information on tissue architecture and micro-environment for the generation of sub-cellular data. Current bulk and scRNA-seq methods provide users with highly detailed data regarding tissues or cell populations but do not capture spatial information7,9,10.
RNA-seq analysis work-flow
1) Experimental design
There are many various library types in RNA-seq resulted in sequencing reads (sequenced transcripts) with different characteristics. For instance, reads can be single-end in which a transcript is read from its only an end (5’ or 3’), however, in the paired-end libraries, a transcript is read from both its 5’ and 3’ end. Paired-end sequencing can additionally help disambiguate read mappings and is preferred for alternative-exon quantification, fusion transcript detection, particularly when working with poorly annotated transcriptomes7. In addition to that, libraries can be stranded or unstranded. The strandedness for libraries is important to determine which DNA strand reads coming from and it is utilized to assign reads to relevant genes. If strandedness information of libraries is misused, then reads are not assigned to true genes, thus gene expression results gonna be wrong11. Besides, technical replicates can be utilized in this process in which one sample is sequenced more than one by using the same high-throughput platform to increase the elimination of technical bias.
2) Laboratory performance
After RNA extraction from all samples, libraries are prepared for sequencing according to the selected library type. After detection of library type, libraries are sequenced to read depth of 10–30 million reads per sample on a high-throughput platform7.
3) Data analysis
After sequencing has been completed, the starting point for analysis is the data files, which contain base-called sequencing reads, usually in the form of FASTQ. The reads having poor quality in FASTQ files are eliminated before the alignment process in which raw sequences are aligned to a reference genome to find their relevant genes. Each sequence read is converted to one or more genomic coordinates and Sequence Alignment Map (SAM) files containing those coordinates are obtained after alignment process7,12. This process has traditionally been accomplished using distinct alignment tools, such as TopHat13, STAR14, or HISAT15, which rely on a reference genome. The SAM files are converted to Binary Alignment Map (BAM) files for further analyses because of their large size and this process is carried out by using Samtools16. After alignment and file conversation steps, reads (transcripts) quantification across samples is performed by using some tools such as featureCounts17 to obtain expression matrix in which each row corresponds to individual genes, however, each column corresponds to individual samples7. Normalization of transcripts abundance across samples is made by using expression matrix to lessen range-based gene expression differences between samples7,18,19. Normalization methods are shown in (Figure 1)20.
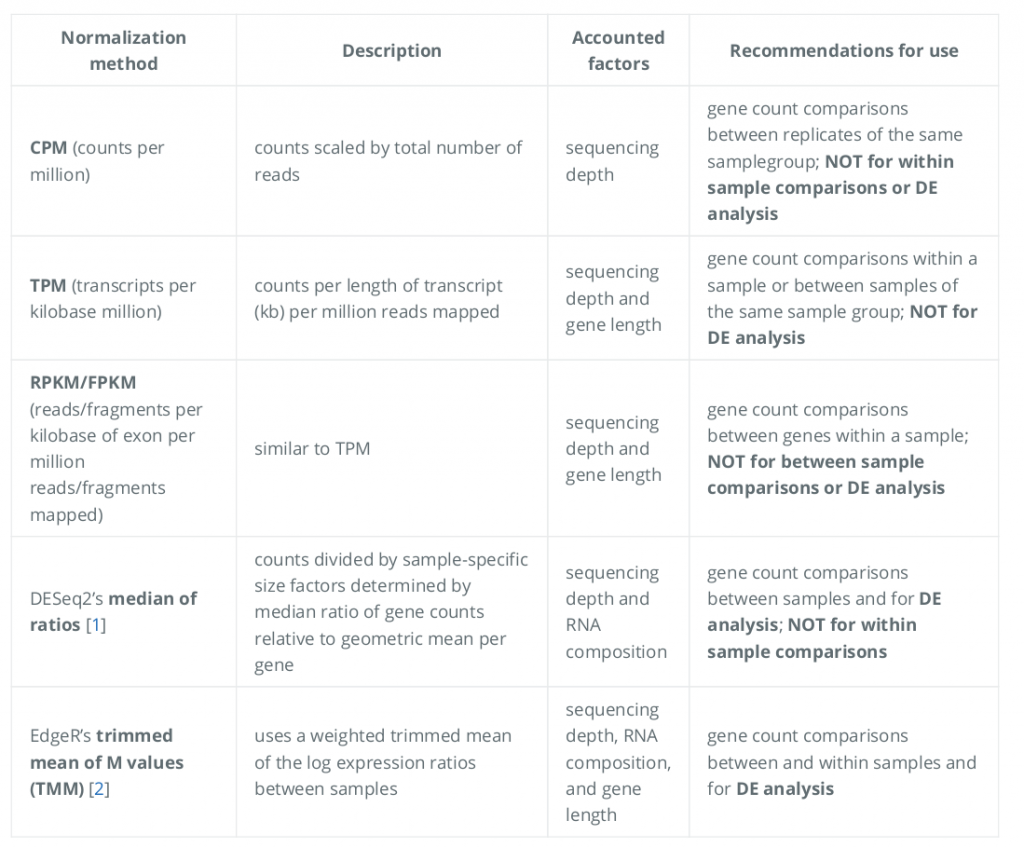
Figure 1. Normalization methods that are used in RNA-seq analyses.
After normalization step, genes with low expression across samples are filtered to prevent statistical noise7, and then statistically meaningful genes (namely, differentially expressed genes) can be detected by using some tools such as edgeR21, DESeq222. In the end, obtained genes can be used for enrichment analyses such as KEGG and Reactome to find out which pathways are affected. RNA-seq technology is utilized for distinct aims, some of which are shown in (Figure 2). The representations of RNA-seq results are shown in (Figure 3).
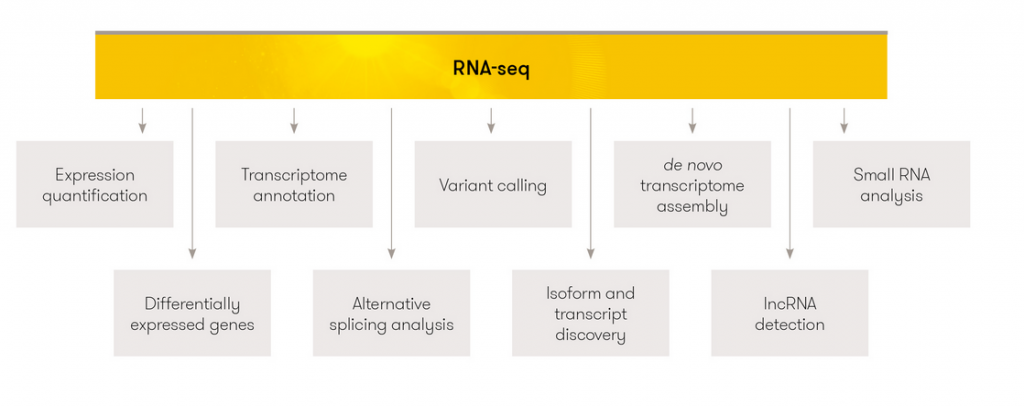
Figure 2. RNA-seq usage fields.
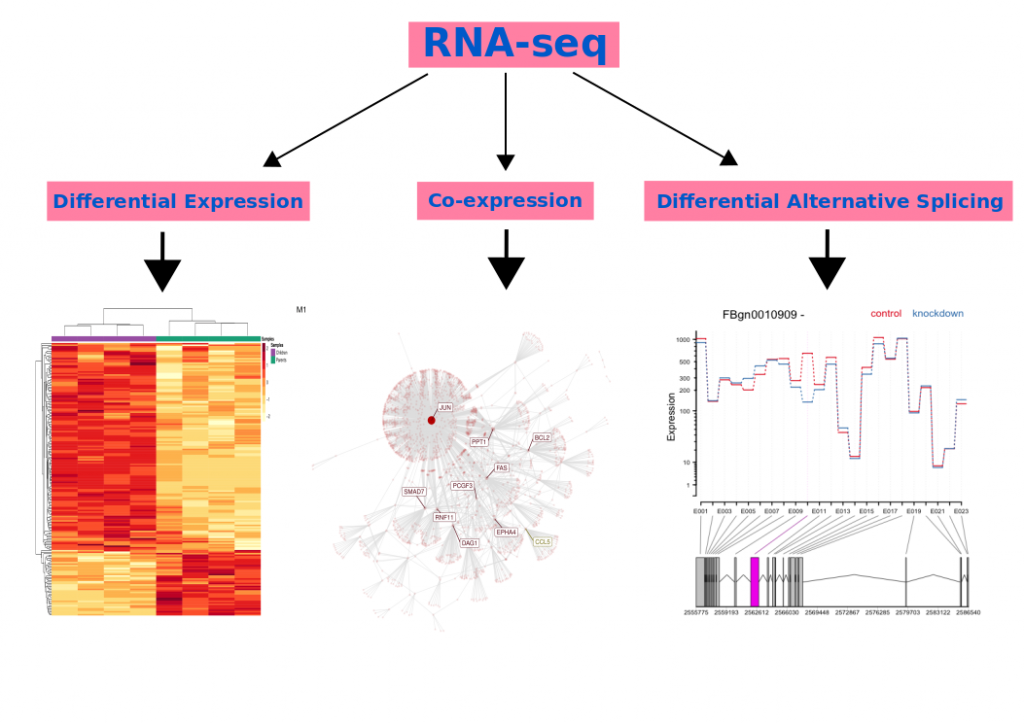
Figure 3. Representation of differential expression, splicing, and co-expression results. In differential expression figure, each row represents the expression amount of a gene, however, each column represents each sample. Red color shows higher expressions, but the yellow color shows lower expressions. In the co-expression figure, a network containing the interaction of each gene with other genes is depicted. In the differential alternative splicing figure, differential usage of E010 exon between control and knockdown groups is depicted.
A detailed RNA-seq work-flow is shown in (Figure 4)12.
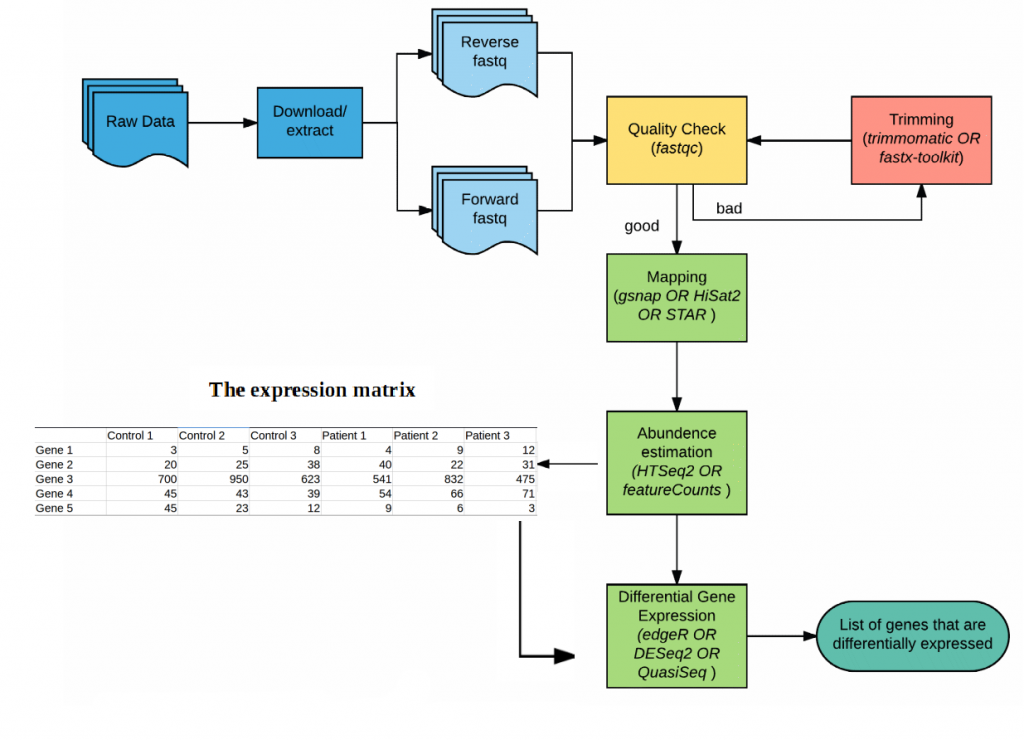
Figure 4. An example of differential expression work-flow.
The various tools that are used for RNA-seq and their tutorials were listed below as well as visualization tools that are used for high-throughput data.
Table 1. List of RNA-seq tool and their usage fields.
| Tool names | Usage | Tutorial Link |
| DESeq222 | Differential expression | https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html |
| edgeR21 | Differential expression | https://bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf |
| DEXSeq23 | Differential splicing | https://bioconductor.org/packages/release/bioc/vignettes/DEXSeq/inst/doc/DEXSeq.html |
| WGCNA24 | Co-expression | https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/ |
| GATK25 | Variant-calling | https://gatk.broadinstitute.org/hc/en-us |
Table 2. List of high-throughput visualization and enrichment tools.
| Tool names | Usage |
| pheatmap26 | Heatmap plot for differentially expressed genes |
| ggplot227 | Most various visualizations ranging from bar charts to violin plots |
| igraph28 | Network visualization for co-expression networks and other network types |
| Enrichr29 | Enrichment analysis of genes |
| DAVID30 | Enrichment analysis of genes |
Note/ Most of the listed tools are dependent on the R statistical computing environment.
Table 3. Examples of differential expression work-flows.
| Examples | Links |
| Example 1 | https://www.bioconductor.org/help/course-materials/2016/CSAMA/lab-3-rnaseq/rnaseq_gene_CSAMA2016.html |
| Example 2 | https://digibio.blogspot.com/2017/11/rna-seq-analysis-hisat2-featurecounts.html |
| Example 3 | https://bioinformaticsworkbook.org/dataAnalysis/RNA-Seq/RNA-SeqIntro/RNAseq-using-a-genome.html |
| Example 4 | https://uclouvain-cbio.github.io/BSS2019/rnaseq_gene_summerschool_belgium_2019.html |
In addition to differential expression pipelines above, If you want to examine my pipeline containing differential expression analysis with DESeq2, you can visit this https://github.com/kaanokay/Differential-Expression-Analysis/blob/master/HISAT2-featureCounts-DESeq2-workflow.md website address in which I attached my Linux and R scripts.
Transcriptome researches in autism spectrum disorder
Autism Spectrum Disorder (ASD) is an early-onset neuropsychiatric disorder. ASD is clinically described with behavioural abnormalities such as restrictive interest and repetitive behaviour. ASD is genetically heterogeneous and heritable (~50%) and 80% of its genetic background is unclear. Aberrations in autistic brains take mostly place in cortex regions (Figure 5) rather than cerebellum. When ASD is compared with other neuropsychiatric disorders such as schizophrenia and bipolar disorder, it has a higher heritability-rate than them, which means that it appears with the more strong genetic background than schizophrenia and bipolar disorder. Studies have revealed that ASD-related genes are enriched in brain-development, neuronal activity, signalling, and transcription regulation. Wnt signalling, synaptic function, and translational regulation are pathways that are affected by mutations in ASD-related genes31.
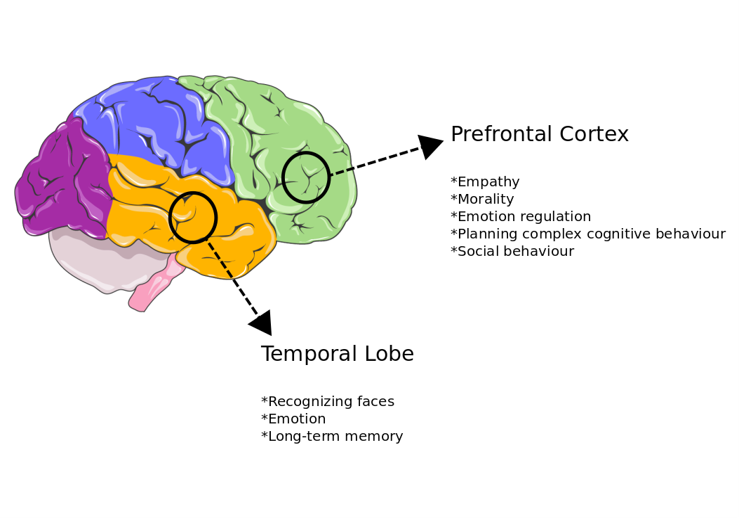
Figure 5. Brain regions most affected in autism.
Transcriptome studies have shown that mRNA, microRNA (miRNA), small nucleolar RNA (snoRNA), and long non-coding RNA (lncRNAs) misexpression occurred in autistic brains. Genes with mRNA misregulation are especially enriched in immune and neuronal pathways, briefly neuronal development and immune system activation are both misregulated in the brains of individuals with ASD. Misregulated miRNAs in autistic brains target mostly genes with synaptic function. Additionally, alternative splicing is misregulated in splicing regulators and this causes mis-splicing patterns in autistic individuals31.
To summarize, RNA-seq is strong technology for understanding diseases and it can be used for various aims.
That’s all 🙂
If you have any questions about this short review and my differential expression pipeline in GitHub, you feel free to contact me via kaan.okay@msfr.ibg.edu.tr e-mail address.
Very thanks for your interest and time!
REFERENCES
1) https://en.wikipedia.org/wiki/Omics.
2) https://en.wikipedia.org/wiki/Transcriptomics_technologies.
3) https://en.wikipedia.org/wiki/Transcriptome.
4) Kadakkuzha, B. M., Liu, X. an, Swarnkar, S. & Chen, Y. Genomic and proteomic mechanisms and models in toxicity and safety evaluation of nutraceuticals. in Nutraceuticals: Efficacy, Safety and Toxicity 227–237 (Elsevier Inc., 2016). doi:10.1016/B978-0-12-802147-7.00018-8.
5) Behjati, S. & Tarpey, P. S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. Ed. 98, 236–238 (2013).
7) Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656 (2019).
8) https://en.wikipedia.org/wiki/Single_cell_sequencing.
9) https://www.10xgenomics.com/spatial-transcriptomics/.
10) https://www.diva-portal.org/smash/get/diva2:1068517/FULLTEXT01.pdf.
11) https://salmon.readthedocs.io/en/latest/library_type.html.
12) https://bioinformaticsworkbook.org/dataAnalysis/RNA-Seq/RNA-SeqIntro/RNAseq-using-a-genome.html.
13) Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
14) Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
15) Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
16) Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
17) Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
18) Evans, C., Hardin, J. & Stoebel, D. M. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief. Bioinform. 19, 776–792 (2018).
19) Liu, X. et al. Normalization Methods for the Analysis of Unbalanced Transcriptome Data: A Review. Front. Bioeng. Biotechnol. 7, 1–11 (2019).
20) https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html.
21) Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
22) Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, (2014).
23) Anders, S., Reyes, A. & Huber, W. Detecting differential usage of exons from RNA-Seq data. Nat. Preced. 1–30 (2012) doi:10.1038/npre.2012.6837.2.
24) Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, (2008).
25) McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
26) https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf.
27) https://cran.r-project.org/web/packages/ggplot2/ggplot2.pdf.
28) https://cran.r-project.org/web/packages/igraph/igraph.pdf.
29) https://amp.pharm.mssm.edu/Enrichr/.
30) https://david.ncifcrf.gov/.
31) Quesnel-Vallières, M., Weatheritt, R. J., Cordes, S. P. & Blencowe, B. J. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet. 20, 51–63 (2019).