
Molecular Docking, Newton dinamiklerini, termodinamiksel ve kimyasal hesaplamaları kullanarak iki molekülün (Reseptör-Ligand) kimyasal ve fiziksel özelliklerine bağlı olarak oluşturabileceği en stabil yapıyı bulabilmek için, yapısal biyoloji alanında çok sık kullanılan bir yöntemdir. Moleküler Kenetlenme, hücre içinde bulunan komplekslerin (protein-protein, protein-ligand protein-antibody) anlık bir görüntüsünü bilgisayar ortamında oluşturup inceleyebilmemize olanak sağlar. Bu çalışmalar ise ilaç geliştirme çalışmalarından bir proteinin mekanizmasının anlaşılabilmesine, yeni bir ilaç adayı molekülün bulunmasına kadar birçok alanda bize bilgi sağlamaktadır. Ligand kimyası (iyonizasyon ve yapısal farklılıklar), Reseptörün esnekliği, proteinin kompleks oluştururken içinde bulunabileceği farklı konformasyonlar ve docking programlarının hesaplamada kullandığı farklı skorlama yöntemlerinin yeterliliği ise molecular docking çalışmalarında karşılaşılan güncel sorunlardır, gün geçtikçe bu problemleri çözmek için yeni algoritmalar ve farklı metotlar geliştirilmekte ve molecular docking çalışmaları her geçen gün daha güvenilir sonuçlar vermektedir (Guedes et al., 2018).
Şimdi bir docking çalışması ile metodumuzu daha iyi anlamaya çalışalım!
AutoDock4.2 ile Docking Çalışması
Aşağıda yapacağım uygulamalı anlatımda genel olarak docking işleminin nasıl gerçekleştiği fazla detaya girmeden genel hatları ile anlatmaya çalışacağım. Detaylı bilgileri merak edecek olanlar için aralıklarla linkler bırakacağım, bu linklerden makalelere gidip detaylı okumalar yapabilirsiniz.
Öncelikle biraz kullanacağımız programdan bahsedelim. AutoDock4 yaygın olarak kullanılan, Linux ve Windows gibi OS’larda çalışabilen ücretsiz bir şekilde indirip kullanabileceğiniz bir docking programıdır. AutoDock4 deneysel (experimental) ve bilgi temelli (knowladge-based) hibrit bir skor fonksiyonu kullanmaktadır. Bu hibrit fonksiyon sayesinde Autodock4, ligandların in-vitro bağlanlanma enerjilerine yakın değerler vermektedir.
AutoDock4.2 Skor Fonksiyonu (Hill & Reilly, 2015);
ΔGbinding = ΔGvdW + ΔGelec + ΔGhbond + ΔGdesolv + ΔGtors
ΔGvdW = 12-6 Lennard-Jones potential (with 0.5 Å smoothing)
ΔGelec = with Solmajer & Mehler distance-dependent dielectric
ΔGhbond = 12-10 H-bonding Potential with Goodford Directionality
ΔGdesolv = Charge-dependent variant of Stouten Pairwise Atomic Solvation Parameters
ΔGtors = Number of rotatable bonds
Program yukarıdaki metotlardan gelen deneysel ve teorik değerleri kullanarak protein ve ligandın atomlarının aralarında yaptığı etkileşimlere enerji olarak değerler verip, kompleks hakkında anlamlı sonuçlar çıkartmamıza yardımcı oluyor.
Şimdi az çok programın nasıl çalıştığı anladıysak uygulamalı bir örneğe geçelim.
Histon Deasetilaz 6 (HDAC6) – Quisinostat
Quisinostat hala deneysel aşamada olan bir HDAC6 inhibitörüdür. Bu çalışmada daha önce ΔG binding ve Ki değeri deneysel olarak hesaplanmış ve Ki = 16.9nM olarak bulunmuştur (Carrillo et al., 2015). Bizde Autodock4 kullanarak bu değere yakın bir sonuç bularak deneysel olarak bulunan değere yakın bir değer bulmaya çalışacağız.
Protein ve Ligandın hazırlanması.
HDAC6 proteinini PDB:5EDU kodu ile Protein Data Bank (PDB)’dan, Quisinostat molekülünü ise pubchem adlı siteden sdf olarak indirebilirsiniz.
Protein ve Ligandın hazırlanması için çeşitli programlar kullanılabilir, bu çalışmada Laboratuvarımızda kullandığımız Biovia Discovery Studio 2016 programını kullanacağız. Bu program çeşitli protokoller için lisans gerektiren bir programdır ama aynı işlemleri Chimera veya PyMol ile de yapabilirsiniz.
Protein ve ligand hazırlamak için neler yapılmalıdır?
PDB’den indireceğimiz yapılar genellikle X-ray kristolografi yapıları oluyor ve bu yapılardan 3D yapı oluşturulurken loop veya proteinin diğer bölgelerinden eksik kısımlar, kristalleştirme sırasında kullanılan proteine ait olmayan moleküller gibi docking işlemi esnasında istemeyeceğimiz, sonucumuzu olumsuz etkileyecek yapıları tamamlamak veya temizlemek için proteini hazırlamamız gerekiyor. Bu işlem sırasında;
- Proteinin yapısına dahil olmayan moleküller siliniyor
- Side-Chain grupları optimize ediliyor
- Su molekülleri siliniyor
- Eksik loop ve diğer bölgeler tamamlanıyor.
- Proteinin fizyolojik koşullarda bulunduğu pH değerine göre side chainler protone ediliyor.
Ligand molekülünü hazırlamak için liganda göre farklı değişikliklerde yapılabileceği gibi, genel olarak protein ile aynı pH değerinde molekülü protone etmek yeterli olacaktır. Eğer bu işlemleri yapacak programa ve imkanlara herhangi bir sebepten erişme imkânınız yok ise buradaki linkten benim hazırladığım ve dockingde kullandığım hazırlanmış protein ve molekülü de indirebilirsiniz.
Bu iki molekülü hazırladıktan sonra bu moleküllerin AutoDock4 programında tanınması için PDB veya diğer desteklenen formatlardan kaydetmemiz gerekiyor.
Proteini ve ligandımızı hazırladığımıza göre şimdi ligand molekülümüzü nereye dock ediceğimizi tespit etmemiz gerekiyor. Bunun için daha önce yayınlanmış makalelerden araştırma yapabilirsiniz ya da kristal yapı oluşturulurken kullanınan (5EDU için) proteinin aktif bölgesinde bulunan TRICHOSTATIN A molekülünün ağırlık merkezini veya aktif side kısmında bulunan ve mekanizma için çok önemli olan Zn+2 atomuna yakın bir bölge seçebilirsiniz.
AutoDock4 ve Docking İşlemleri
AutoDock4 programının docking işlemleri için bazı parametre dosyalarına ihtiyacı vardır. Bu dosyaları oluşturmak için yine AutoDock programını geliştiren grup tarafından geliştirilen ADT (Autodock Tools) programını indirmemiz gerekiyor. Bu programı AutoDock4 (docking için) ve Autogrid4 (grid parametrelerini) hazırlamak için kullanacağız. Programları buradan indirebilirsiniz ve kurulumları için talimatları yine aynı siteden bulabilirsiniz.
Şimdi sırası ile Autodock Tools kullanarak gerekli dosyaları hazırlayalım.
1) Protein ve Ligandın Yüklenmesi
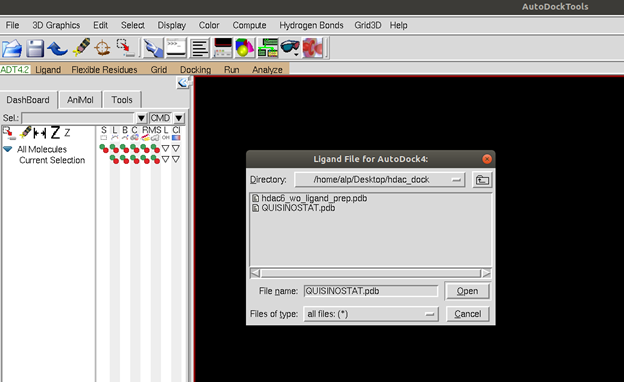
Programı açıp “Ligand –> Input -> Open ->” kısmında ligandımızı pdb olarak seçelim. Bu seçimi onayladıktan sonra program ligand hakkında ekrana bazı bilgiler vericek. Ardından ligandımızı yine aynı sekmedeki output seçeneği ile pdbqt olarak kaydedelim.
Proteinimizi programa yüklemek için “Grid -> Macromolecule -> Open ->” kısmından aynı şekilde proteinimizi pdb formatinda seçelim ve yine output seçeneğini kullanarak pdbqt olarak yazdıralım. Burada HDAC6 açısından dikkat etmemiz gereken bir durum oluştu, program aynı şekilde kaydettikten sonra ekrana bazı uyarılar veriyor ve bağlanma bölesinde kofaktör olan Zinc (çinko) atomunu değersiz olarak kaydediyor fakat HDAC6 içerisinde bu atomun +2 değerlik aldığını biliyoruz. Bu durumda pdbqt klasörüne giderek zinc atomunun değerliğini +2 ile manuel olarak değiştirmemiz gerekiyor.
2) Grid Parametre Dosyalarının Hazırlanması.
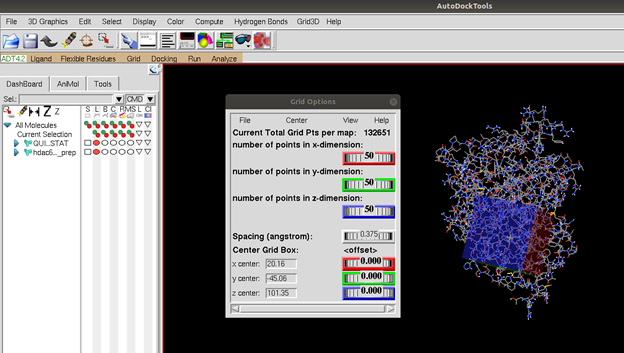
“Grid -> Set Map Types -> Chose Ligand” kısmından ligandı seçtiğimizi belirtiyoruz, ardından “Grid -> GridBox” seçimini yaparak ekrana gelen “grip options” kısmından tekrar “Chose Ligand” seçimini yaparak gireceğimiz koordinatların ligand için geçerli olduğunu belirtiyoruz. Daha sonra x, y, z koordinatlarına, kristal yapıdaki TRICHOSTATIN A molekülünün ağırlık merkezi olarak belirlediğimiz koordinatları yazıyoruz. Burada dikkat etmemiz gereken bir diğer husus ise grid box dediğimiz Figür 2’de mavi ve kırmızı ile görülebilen 3 boyutlu yapının ligandın sığabileceği boyutlarda olması. Bu yapının fazla büyük ya da fazla küçük olması docking sonuçlarınızı etkileyecektir. Docking esnasında ligand bu kutu içerisinde pozisyon aramaktadır ve eğer fazla büyük, fazla küçük veya yanlış yerde belirlenirse sonuçlarınız maalesef olumsuz etkilenecektir. Bu işlemde yapıldıktan sonra “File -> Close Saving Current” seçimi yaparak .gpf (grid parameter file) dosyanızı ligand ve proteinin bulunduğu dosyaya kaydetmeniz gerekiyor.
3) Docking Parametre Dosyalarının Hazırlanması
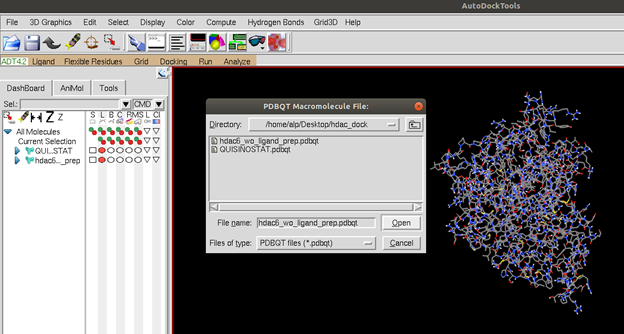
“Docking -> Macromolecule -> Set Rigid Filename” sekmelerinden makro molekülümüzü yani proteinimizi seçiyoruz ve bu seçim aynı zamanda docking esnasında proteinimizi sabitlemeye yarıyor peki neden proteini sabitliyoruz (Figüre 3)? Program çalışırken her bir adımda ligand molekülündeki hareket edebilecek, moleküle esneklik sağlayan ve belli açılara kadar esneyebilen noktalar program tarafından önceden tanımlanmıştır. Program bu noktaları farklı şekillerde, belli açılar içerisinde çevirerek ligand için proteinin içerisinde yeni pozisyonlar arar ve bu pozisyonlar için enerji hesaplamaları yapar. Bundan dolayı bir ligandta bu noktalardan ne kadar çok olursa docking işlemi o kadar uzun sürer. Ligandların aksine proteinde bu noktalardan yüzlerce sayıda olabilir. Bu yüzden zamandan kazanmak ve hesaplama yükünü azaltmak için ligand serbest, protein ise sabit bırakılır. Şimdi iste “Docking -> Ligand -> Chose Ligand” seçeneği ile ligandımızı seçiyoruz.
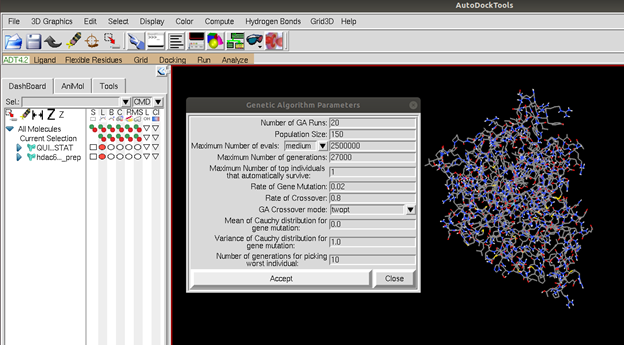
Ligandımızın ve proteinimizin docking esnasında nasıl davranacağını program da belirledikten sonra şimdi docking için bir genetik algoritma belirlemeliyiz. Docking işlemini daha iyi gerçekleştirebilmek ve optimum bağlanma modunu bulabilmek için birçok genetik algoritma geliştirilmiştir. Bu uygulamada Lamarckian Genetik algoritmasını kullanacağız. Genetik algoritma ile ilgili detaylı bilgileri ve Figür 4’teki seçeneklerin ne ifade ettiğini öğrenmek için buradaki makaleyi okuyabilirsiniz. Genetik algoritmayı seçip “accept” diyerek devam ettiğimizde program bize bir .dpf (docking parameter file) oluşturuyor. Bu dosyanın içinde şu ana kadar yaptığımız seçimlerin programın okuyabileceği bir formatta çıktısı var. Bu dosyayı da oluşturduğumuz diğer dosyaların içine kaydediyoruz.
4) Terminalden Docking işleminin başlatılması
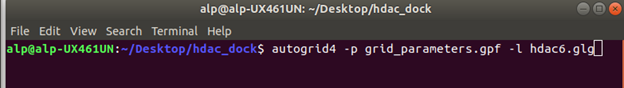
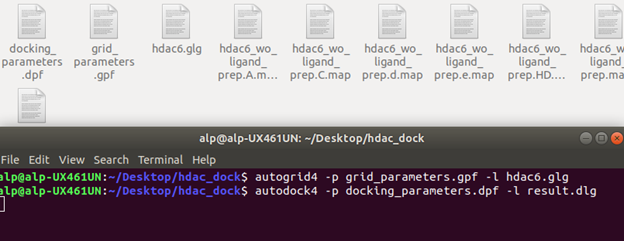
pf ve dpf dosyalarımızı oluşturduğumuza göre Autodock Tools uygulamasını artık kapatabiliriz. Şimdi bu dosyaları kaydettiğimiz dosyada bir terminal açıp, autogrid4 komutunu Figür 5’teki gibi çalıştırıyoruz. Autogrid bize map dosyalarını oluşturuyor bu dosyalar docking esnasında bize gereken dosyalar. Ardından Figür 6’da olduğu gibi şekilde Autodock komutunu çalıştırıyoruz.
Bu işlemden sonra Docking başlamış oluyor, yeni bir terminalde “top” yazarak işlemin çalışma durumunu takip edebilirsiniz.
5) Sonuçlar

İşlem tamamlanınca çalıştığımız klasörün içinde bir .dlg dosyası oluşturuluyor. Bu dosyada docking sonuçlarımız mevcut. Bütün sonuçları enerji cinsinden görmek isterseniz “CLUSTERING HISTOGRAM” başlığı altında bir tabloda bu değerleri görebilirsiniz. Daha detaylı sonuçları için dosyanın alt kısımlarına doğru “LOWEST ENERGY DOCKED CONFORMATION from EACH CLUSTER” olarak hazırlanmış bir kısım var burada karşımıza ilk gelen sonuç programın oluşturduğu protein-ligand kompleksleri içerisinde en düşük enerjiye sahip olan komplekse ait. Burada ΔG ve Ki değerlerini görebiliriz. Yazımızın başında deneysel olarak hesaplanan Ki değerini16.9 nM olarak söylemiştik biz ise burada 89.68 nM olarak bulduk, nanomolar seviyesinde bu değerler yaklaşık değerler olarak kabul edilebilir. Daha detaylı karşılaştırmak için bağlanma enerjilerine de bakabiliriz.
Enerjileri karşılaştırmak için Ki den ΔG’ye geçmemiz gerekiyor, bunun için kullanacağımız formül;
ΔG = -RTlnKi
Bu formülde R = 0.008314 kJ mol-1 K-1 ve T = 298,15 K değerlerini yerleştirirsek deneysel olarak gerçekleştirilen bağlanma enerjisini -7.02 kcal/mol olarak buluyoruz, bizim ise Autodock ile bulduğumuz sonuç -9.61 kcal/mol.
Autodock4 kullanarak deneysel olarak çıkan sonuca yaklaşık bir değer bulduğumuzu söyleyebiliriz. Şunu da unutmayalım bu işlemin laboratuvarda yapılması bir, bir buçuk saat sürmekte ve harcanan malzemeler (enzim, molekül ve assay kitleri) de cabası, biz bu değere yaklaşık bir değeri yaklaşık 5 dakika içerisinde bulabiliyoruz ve bu sadece bir molekül için. Docking çalışmalarının gün geçtikçe daha fazla tercih edilmesinde ki bir nedende bu. Bizler, bilgisayar destekli ilaç tasarımı ile uğraşan insanlar bu yöntem ile bir günde on binlerce molekülü bilgisayar ortamında tarayıp sonuç çıkartabiliyoruz ve bunu yaparken deneysel sonuçlara yakın değerlerde buluyoruz.
Bu konu ile ilgili öğrenilecek daha çok fazla detay ve öğrenilecek çok fazla şey var sizlere bildiğim kadarı ve elimden geldiğince anlatmaya çalıştım, Umarım bu yazı sizler için bilgilendirici olmuştur. Çalışmaya devam! 🙂
Referanslar
- Carrillo, A. K., Guiguemde, W. A., & Guy, R. K. (2015). Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorganic & Medicinal Chemistry, 23(16), 5151–5155. https://doi.org/10.1016/j.bmc.2014.12.066
- Guedes, I. A., Pereira, F. S. S., & Dardenne, L. E. (2018). Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Frontiers in Pharmacology, 9. https://doi.org/10.3389/fphar.2018.01089
- Hill, A. D., & Reilly, P. J. (2015). Scoring Functions for AutoDock (pp. 467–474). https://doi.org/10.1007/978-1-4939-2343-4_27