
Structures of biological macromolecules cannot be easily determined, as they are flexible, i.e. their conformations change while they function [1]. Therefore, these molecules should be characterized. This characterization step might be very challenging [2]. Structures of these macromolecules can be specified by using some well-known techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and Small-angle X-ray scattering (SAXS) [3]. These techniques give different information about structure of a molecule and these alone are not enough to determine whole structure properties [4]. Also, results of these techniques need to be interpreted by using computational analysis in order to specify more precise structures of macromolecules [2]. Integrative modeling is a common technique used in recent years to more accurately determine the molecular structure.
What is integrative modeling?
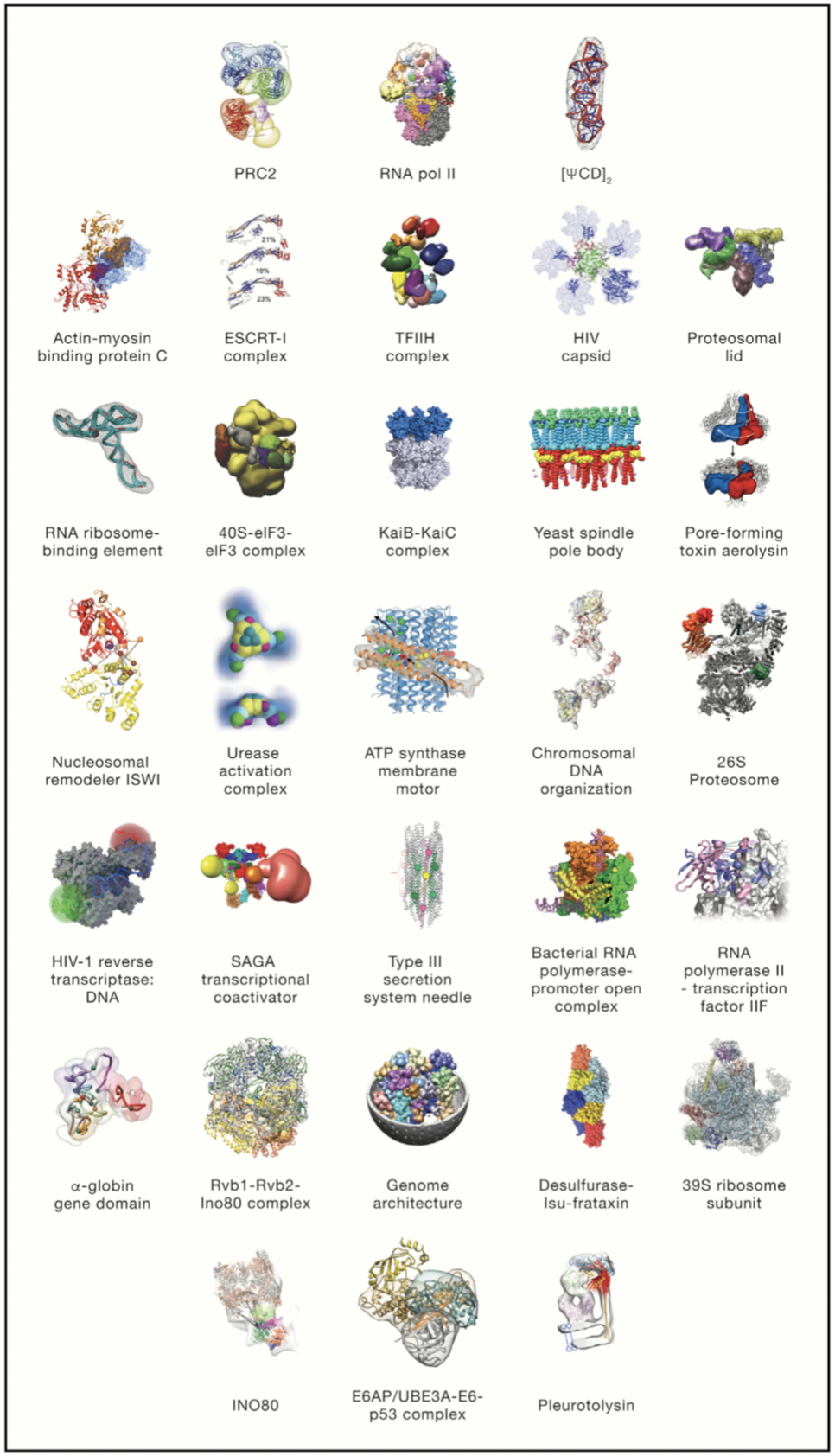
As understood from a word meaning, integrative modeling uses more than one information source to model a structure and mechanism of biological molecules in systems [5]. As in all modeling methods, integrative modeling combines all available experimental data as well as with computational techniques to obtain more accurate, precise, complete and efficient model (Figure 1).
Integrative modeling has iterative four stages: (1) gathering information, (2) representing the system by translating information, (3) creating sample of structural models and (4) scoring the model (Figure 2).
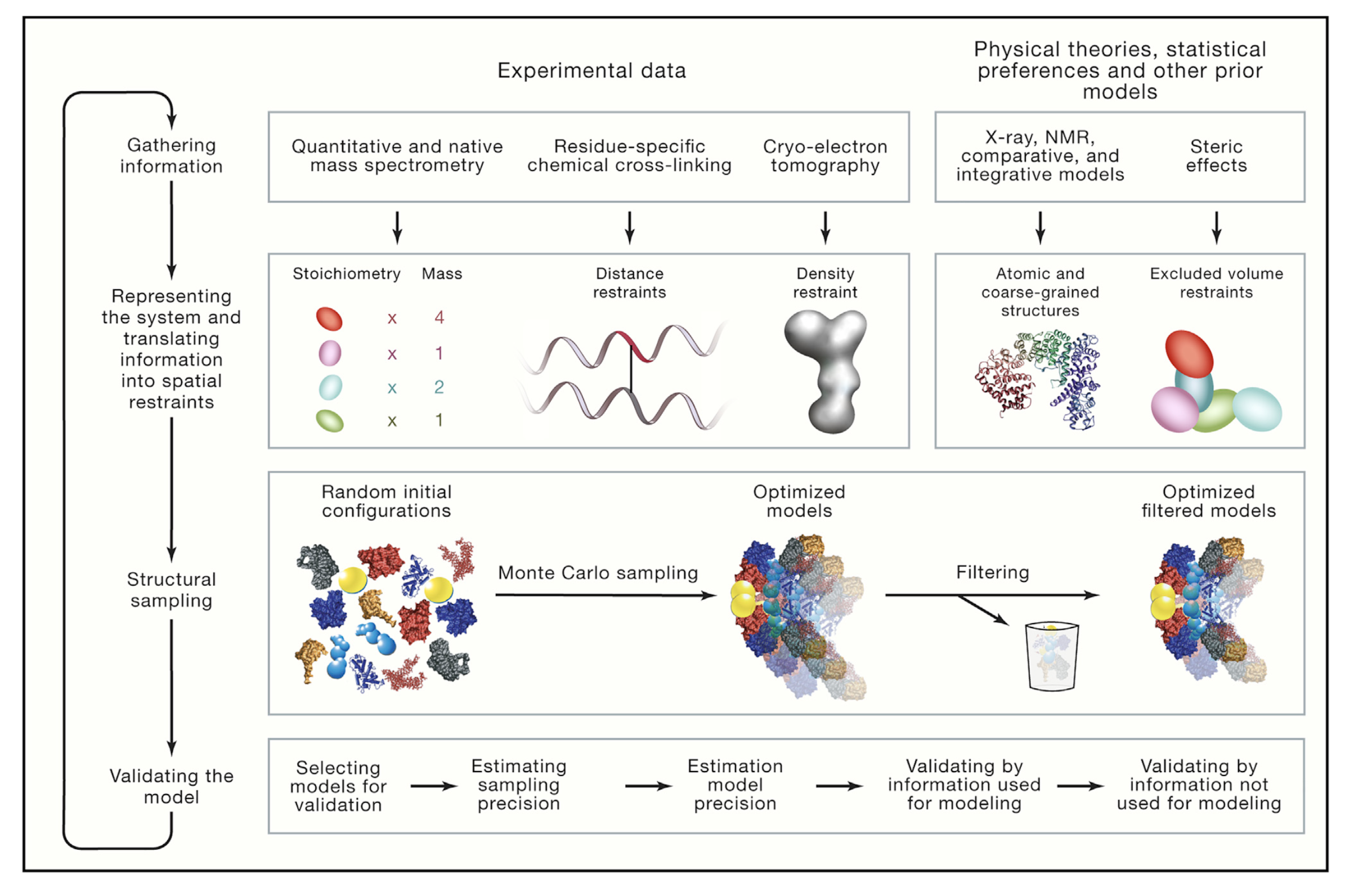
Briefly:
- Gathering information: Collecting all available data about structure of the system is the first stage in Figure 2 [6]. Structural information coming from any method/technique can be based in the theory.
- Representing the system and translating information into spatial restraints: Gained information data from first step can be used to describe a model [5]. Based on the input information, variables are used to define features of the model. These variables can represent atoms, coarse-grained particles and subunit in complex in structural biology.
- Structural sampling: Created models have random configuration as a first. Then, different configurations are sampled based on the scoring functions [6]. Results are fitted to input information for filtering.
- Validating the model: Models which have good-scoring results are chosen for validation (these chosen models creates ensemble) [5]. After some estimation, one or more than one model can be chosen as a result (or not to be chosen). This depends on accuracy calculation based on the input data.
As a following, you can find another representation of these iterative four stages for integrative modeling which shows different usage areas [7].
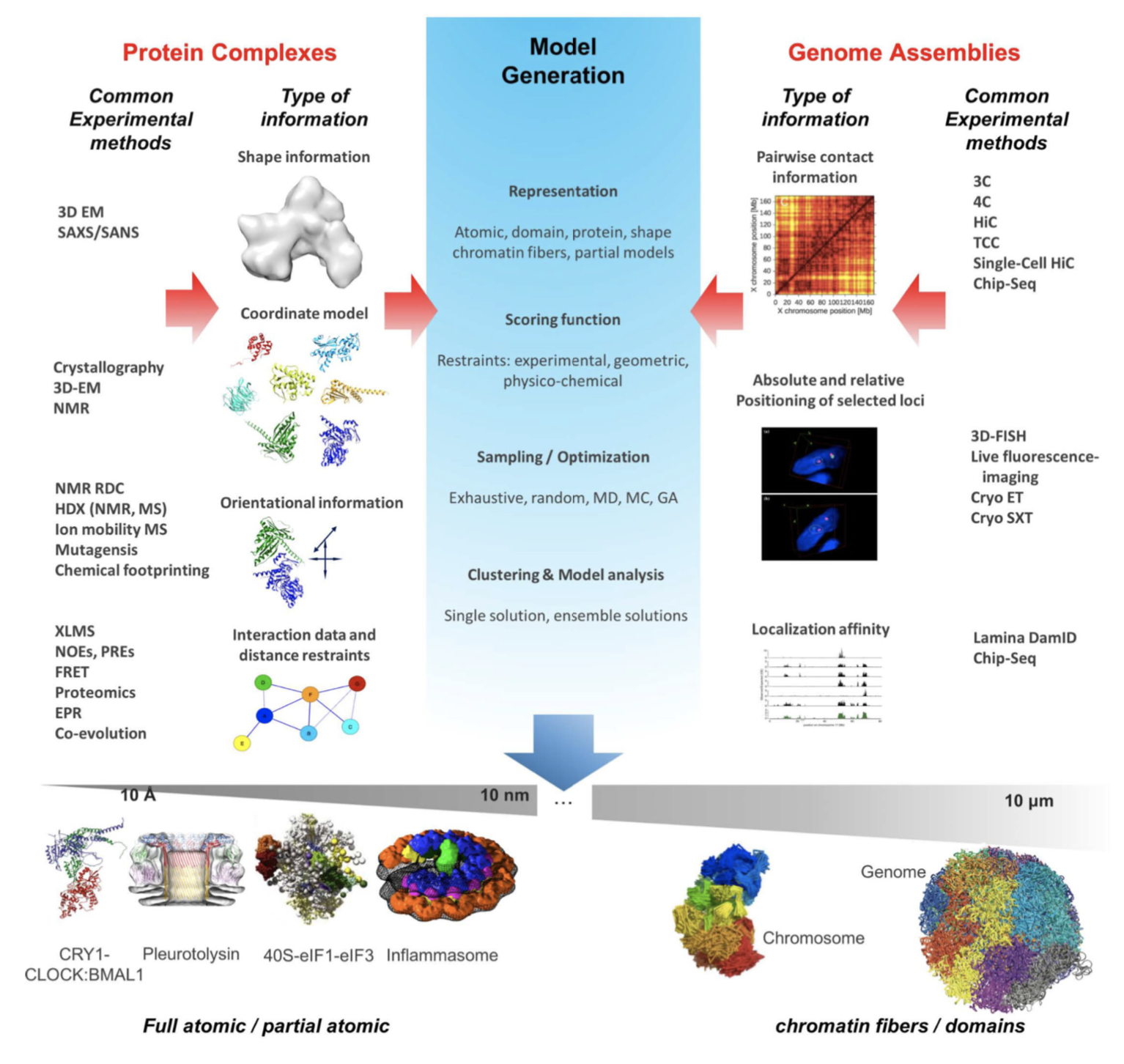
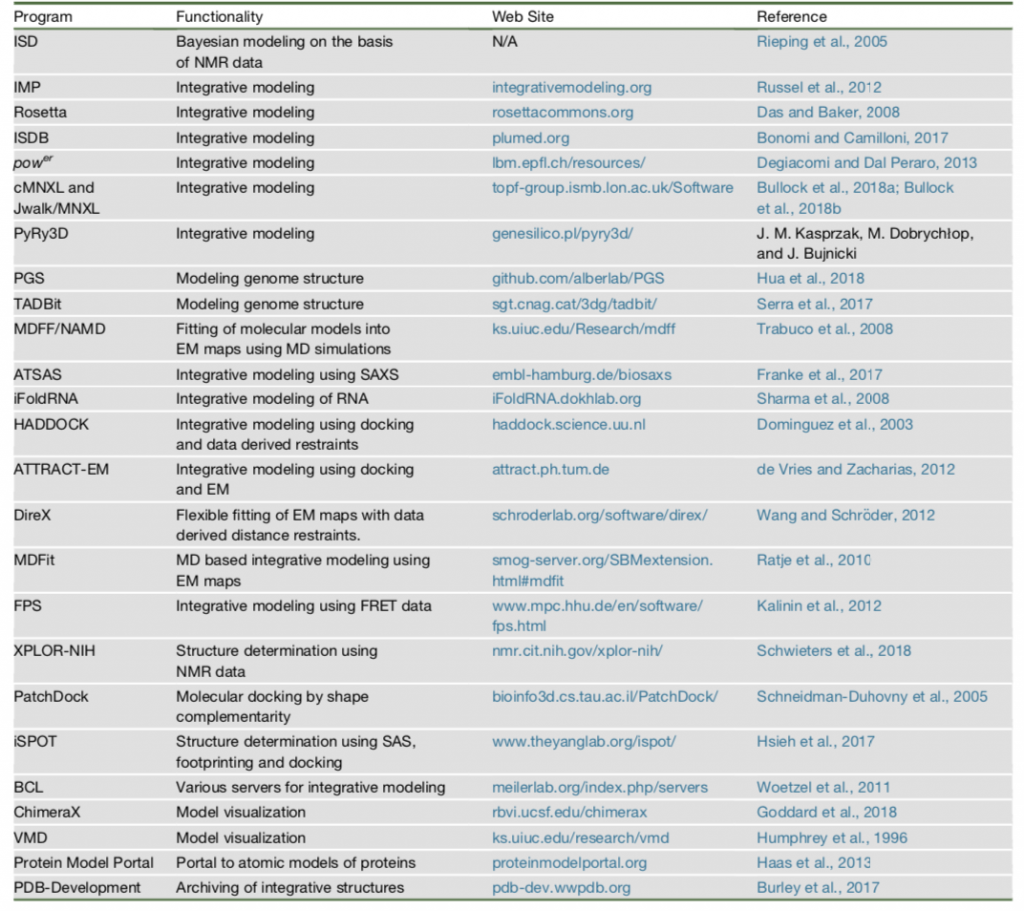
Types of Structural Information & Software Resources for Integrative Modeling
X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy are common techniques to specify the structure of biological molecules[4]. They give atomic information of the structure. Acquiring crystallographic structures is very challenging for large biological complexes. Therefore, some recent techniques to determine structure of a molecule are used as shown in Table 1.
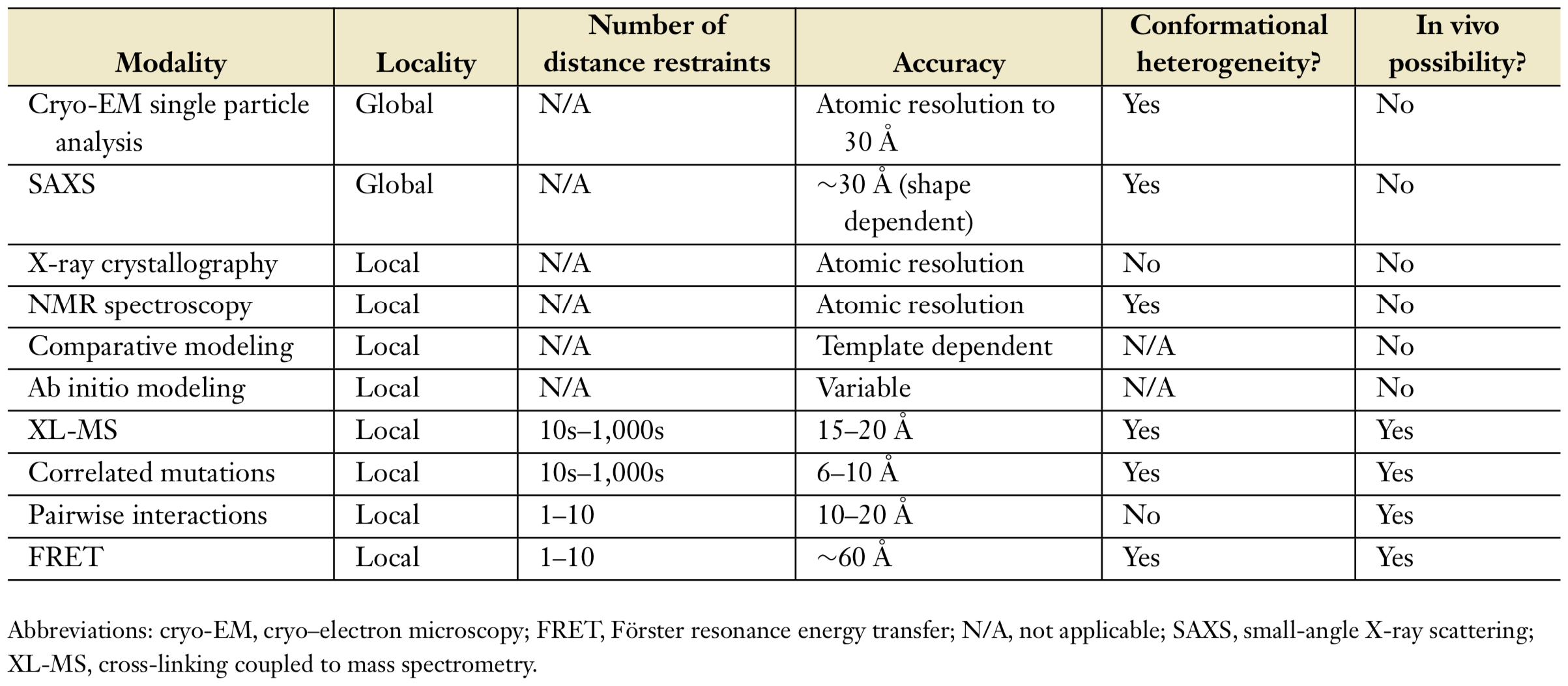
Each technique gives a different aspect about structure of a molecule, so they can be used to determine different properties of the structure[5]. Brief explanation of some techniques in Table1:
- Cryo-EM: Cryo-electron microscopy is commonly used to specify global structural information and structural characterization of large complexes [4]. The three-dimensional (3D) electron density map of a macromolecule complex is obtained with cryo-EM single particle analysis.
- XL-MS: Distance restraints between residue pairs in biological molecule can be determined by using cross-linking coupled to mass spectrometry.
- SAXS: Small-angle X-ray scattering is commonly used method to get information about shapes of macromolecules.
- Sequence information is important to get evolutionary conserved positions which can be related with folding, function, interactions and dynamics of the molecule.
- FRET: Förster resonance energy transfer is used to get information about structures, dynamics and interactions of protein.
In Table 2, some of commonly used suite of tools with their methods are listed which were also used to create structures in Figure 1.
What is the accuracy of integrative modeling?
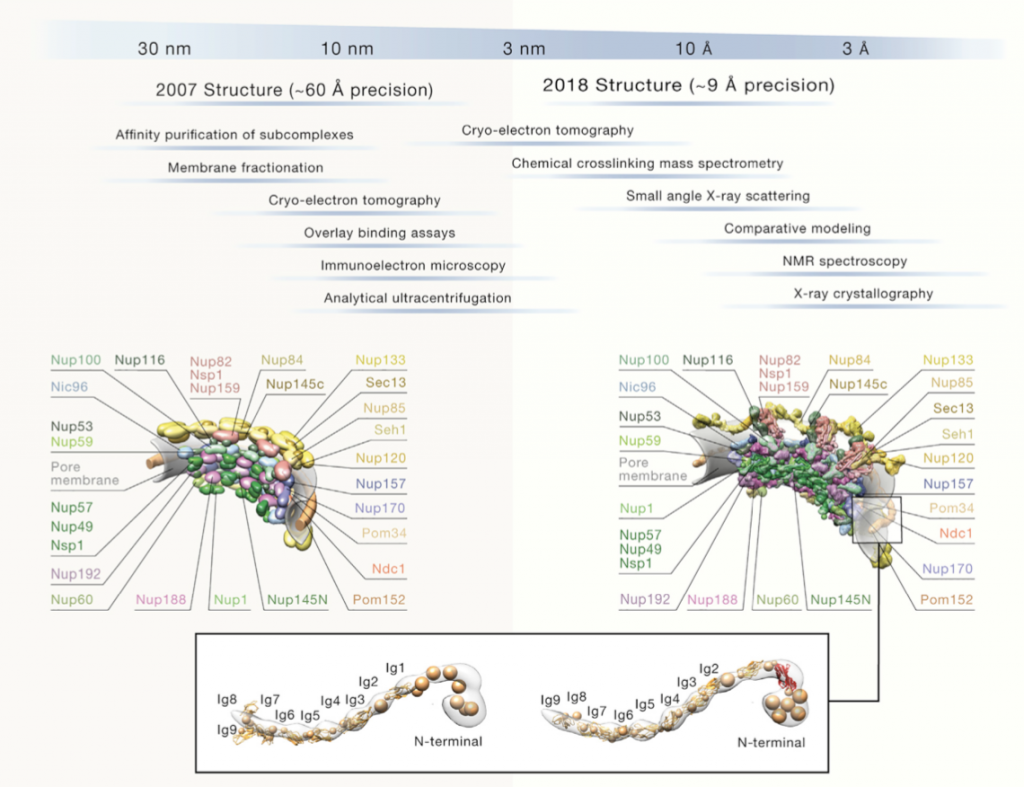
The modeled structures of the Yeast NPC from two different years are compared in Figure 4 [5]. As it can be seen in Figure 4, 2018 structure of Yeast NPC was modeled more detailed than 2007 structure. Among the years, precision is getting smaller and that leads to more detailed, more accurate model. Accordingly, decrease on the value of precision means increasing the resolution. High resolution allows us to determine structure more clearly. Therefore, 2018 structure has more details. These two structures of the Yeast NPC were not modeled by using only one information resource. They were modeled with multiple information from different resources. The resources used for 2018 model give more detailed information. Purpose of integrative modeling is to reach as true as possible model by using all available data and the quality of data increases the accuracy of integrative modeling. Figure 4 is a good example of how well integrative modeling works and how used resources affect the result.
——————————————————————————————————————————————————————
I suggest you take a look at the articles I used for this short review. “Principles for Integrative Structural Biology Studies” article is very informative review for integrative modeling. If you will choose only one, I highly recommend that article. I hope this short review is helpful for you. Thank you for reading.
——————————————————————————————————————————————————————
References
[1] A. Panjkovich and D. I. Svergun, “Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis,” Phys. Chem. Chem. Phys., vol. 18, no. 8, pp. 5707–5719, 2016.
[2] E. Karaca and A. M. J. J. Bonvin, “Advances in integrative modeling of biomolecular complexes,” Methods, vol. 59, no. 3, pp. 372–381, 2013.
[3] C. E. M. Schindler, S. J. de Vries, A. Sasse, and M. Zacharias, “SAXS Data Alone can Generate High-Quality Models of Protein-Protein Complexes,” Structure, vol. 24, no. 8, pp. 1387–1397, 2016.
[4] M. Braitbard, D. Schneidman-Duhovny, and N. Kalisman, “Integrative Structure Modeling: Overview and Assessment,” Annu. Rev. Biochem., vol. 88, no. 1, pp. 113–135, Jun. 2019.
[5] M. P. Rout and A. Sali, “Principles for Integrative Structural Biology Studies,” Cell, vol. 177, no. 6, pp. 1384–1403, 2019.
[6] B. Webb et al., “Integrative structure modeling with the Integrative Modeling Platform,” Protein Sci., vol. 27, no. 1, pp. 245–258, Jan. 2018.
[7] A. P. Joseph, G. Polles, F. Alber, and M. Topf, “Integrative modelling of cellular assemblies,” Curr. Opin. Struct. Biol., vol. 46, pp. 102–109, 2017.