
Moleküler Dinamik Simülasyon Nedir ?
Moleküler dinamik (MD) simülasyonları, kabaca tabir etmek gerekirse, temeli klasik mekanik olarak da adlandırılan Newton’nun hareket kanunlarını kullanarak (Newton’s laws of motion) bir protein veya molekülün atomlarının zamana göre hareketini tahmin eder. Gününümüzde çok sıklıkla başvurulan bir yöntem olmasına rağmen geçmişi 1950’lere kadar dayanıyor. 1950’lerin sonunda, B. J. Alder and T. E. Wainwright tarafından ilk MD simülasyon basit gazlar üzerinde denenmiştir. Sonrasında ise 1977 sonlarında ilk defa proteinler üzerinde çalışılmaya başlanmıştır. Günümüzde bu simülasyonların kullanımını daha çok mümkün kılan üç isim Martin Karplus, Michael Levitt and Arieh Warshel “for the development of multiscale models for complex chemical systems.” çalışmalarından dolayı 2013 Nobel Kimya ödülüne layık görülmüştür (bknz. The Nobel Prize in Chemistry 2013).
Peki neden biz bu tür bir çalışmaya ihtiyaç duyduk ? Buna bir sürü yanıt verilebilir ama basit bir cevap verecek olursak. Proteinlerin 3 boyutlu yapılarının elde edildiği X-ray kristalografi, NMR veya Cryo-em gibi yöntemlerin, hücre içi reaksiyonların çok hızlı gerçekleşemsinden kaynaklı olarak, belirli deneysel metodlarla proteinlerin reaksiyon öncesi ve sonrasındaki yapıları elde edebiliyor olması ve reaksiyon için gerekli olan konformasyonel değişimleri yakalamıyor olması, bizim bazen reaksiyonlar ve hücre içi iç dinamiğin nasıl işlediği hakkında soru işaretleri meydana getiriyor. Tabiki de bu durum proteinden proteine farklılık göstermektedir. Ayrıca bu çalışmalarının zahmetli ve uzun deneyler gerektiriyor. Ufa bir örnek verecek olursak, GTP hidrolizini yapan proteinlerin (aynı zamanda GTPase de denir), reaksiyon için gerekli olan konformasyonu, hidroliz olmayan GTP kullanarak elde edilir. Burada elde edilen konfromasyonun gerçekten de reaksiyon için gerekli olan konformasyon olup olmadığı tartişmaya açıktır. Bu yüzden çok sık tartışmaların döndüğü örnekler literatürde vardır. MD kullanımını neden gerekli olduğunu anlatmak için bir sürü örnek verilebilinir ama basit bir örnek olarak bu verilebilir.
MD için biraz daha bilgi vermek gerekirse, MD sistemleri hızlıdır ve parametriktir. Yani sistemdeki atomlar ve atomlar arsındaki bağlar belirli parametrelere göre tanımlanır. Aynı zamanda toplam enerji korunur. MD’de kovelent bağ kırılması veya oluşumu gözlenmez. Atomları tanımlayan parametreler de genel kimya ve fizikten de bilenen 5 temel enerji hesabı ile tanımlanır. Bunlar bağ gerilimi (bond stretching), açı bükülümü (angle bending), torsion dedimiz dönmeler, elektrostatik kuvvetler ve van der Waals kuvvetlerdir. Bu parametreleri de içeren atomların MD için kullanılacak özelliklerini içeren verilerin bütününe de force-field adı da veriliyor ve birden fazla force-field bulunabiliyor örneğin AMBER force-field, CHARMM force-filed. Çalışmalara göre uygun olanı kullanılabilir.
MD simulasyonun temel işleyişi sırasıyla şöyledir. İlk olarka sistemin potansiyel enerjisi ilgili force-field kullanılarak hesaplanır. Bu enerjinin pozisyona göre türevi, o pozisyondaki kuvveti elde etmemize yarar. Bu kuvvetten de Newton mekaniğinden ivmeye, ivmeden de belli bir zaman aralığındaki hız değişimine gidilir. Bu hız değişiminden de yeni pozisyona gidilir. Sonuç olarak dt kadar bir zaman aralığındaki hız ve pozisyon değişimi hesaplanmış olur. Aşağıda işlemlerin matametiksel ifadelerini sırasıyla bulabilirsiniz.
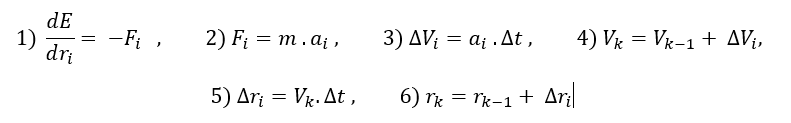
Şimdi yukarıdaki işlemler MD temel mantığı ve nasıl ilerlediğidir. Bizim n atomlu bir sistemimizde 3n tane kordinatımız olacak. Bundan dolayı analitik çözüm pek mümün değildir ve nümerik çözüm uygulanır. Bunun için de “time symmetric” integrasyon metoduları dediğimiz metodlar kullanılır örneğin Leapfrog ve Verlet. Bunların ayrıntılı bilgisi için Andrew R Leach – Molecular modelling principles and applications (2001) kitabını inceleye bilirsiniz.
AMBER’de MD Simülasyon Örneği
- Pdb dosyasının hazırlanması
Burada basit bir şekilde bir Protein Data Bank üzerinden alınan bir protein için simülasyon basit başlatırken neler yapmalıyız ona bakacağız. Bunun için ise kullanacağımız PDB kodu 1L1D (https://www.rcsb.org/structure/1l1d). İlk olarak kullanılan proteinin aktif halinin monomer mi dimer mi veya türevleri şeklinde çalışılıp çalışılmadığının bilinmesi gerekiyor. Çalışacağınız protein hakkında ne kadar bilginiz olursa Moleküler Dinamik Simülasyonunu o kadar iyi uygulayabilirsiniz. Pdb formatındaki yapı -bizim şuan kullanacağımız yapı gibi- X-ray kristalografiden elde edilen tekrarlı yapılar olabilir. Bu yüzden resim 1’de göründüğü gibi yapıdan fazladan chain görülebilir. Bu chaine ait olan her şeyin silinmesi gerekiyor. Bunu text editör yardımı ile veya pymol gibi programlar kullanarak yapabilirsiniz. Bizim sistemimiz için B chainini ve onun içerdiği her şeyi sildik. Ayrıca sistemde kristalografi de kullanılan bufferdan kalan moleküller olabilir bunların da silinmesi gerkiyor. Dikkat edilmesi gerekilen bir husus, bir amino asit zincirinin bitiğini ve yeni bir yapının başladığını -bu yeni bir chain, su molekülleri ya da bir ligand olabilir- AMBER’in anlayabilmsi için PDB dosyasında ilgili chainin sonunda “TER” bulunması gerek. Ayrıca yapıda olmayan amino asitler varsa bunun homoloji model veya abinitio gibi yöntemlerle modellenmesi gerekmektedir. Bizim sistemimiz için böyle bir şeye ihtiyaç yok ve kristal yapıda eğer su varsa bunların tutulmasında yarar vardır. Pdb dosyasın en sonunda bazen “CONNECT” ile başlayan bağları tanımlayan ifadeler bazen AMBER için yanlışlıklara neden olabileceğinden silmeliyiz. Sonuç olarak pdb dosyamızın içinde sadece olmasını istediğimiz yapıların kalmasını istiyoruz ve gerisini siliyoruz. Bizim sistemimiz için bunlar su molekülleri ve proteinin A zinciri.
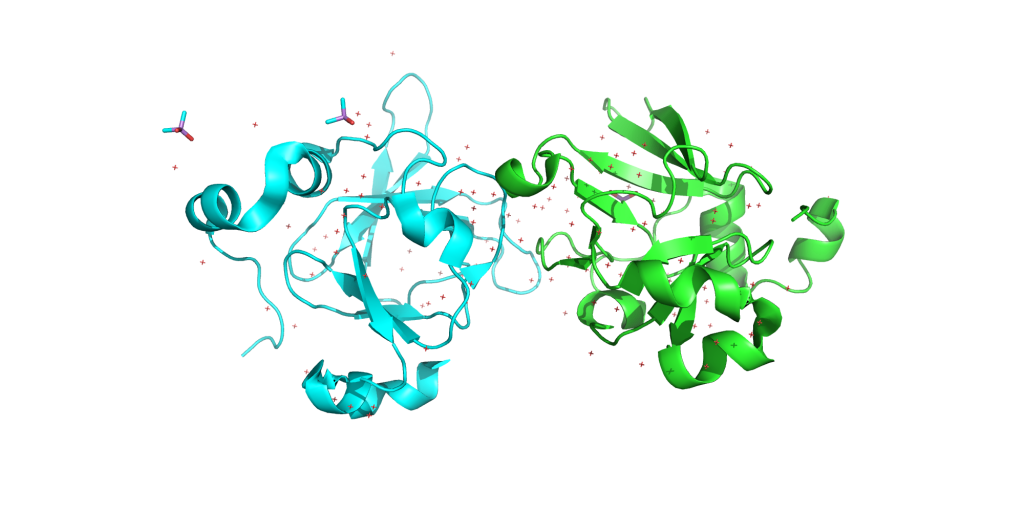
Resim 1. 1L1D yapısının görüntüsü.
Bunun yanında, sistemde standart olmayan amino asitler varsa ve eğer bu standart olmayan amino asitler sistemimiz de olamsını istediğimiz bir durum ise parametresinin eklenmesi gerekiyor. Aynı zaman da bir ligand ile çalışma yapacağımız zaman da parametrelerinin eklenmesi gerekecek. Bunun için çeşitli yöntemler var (bknz. CGenFF). Eğer istenmeyen bir durum ise bunu standart haline çevirmemiz gerekmektedir. Bizim proteinimizde metiyoninler, selenometiyonin olarak gözüküyor. Burada bir text editör yardımıyla pdb içinde “MSE” olarak yazılmış amino asitleri metiyonin AMBER için standart ismi olan “MET” ile değiştirmemiz ve selenyum içeren atomunun olduğu satırı silmeliyiz. Burada sildiğimiz atomun yerine sonradan AMBER kendisi kükürt atomunu koyabildiğinden yerine bir şey yazmamız gerekmiyor. Aşağıda bir örnek verilmiştir. Bu işlemi bütün “MSE” olan amino asitlere yapmamız gerekiyor.

- Protonasyonların Tanımlanması
Bundan sonraki aşamada ise bazı iyonize olabilecek amino asitlerin protonasyonlarının düzenlenmesi olacak. Bu kısım biraz uzun ve zahmetli olabiliyor. Bu yüzden otomatik yapan programlar mevcut örneğin Charmm-gui, fakat burada uzun anlatımını yapacağız. Protonasyonu belirlememiz için iyonize olabilecek amino asitlerin pKa değerlerinin hesaplanması gerekiyor. Bunun için PROPKA kullanacağız. Probka, pKa’ı ilgili amino asitin etrafındaki etkileşimlerine ve solvent erişebilir bir bölge olup olmama durmuna göre bize bir tahmin değer çıkarıyor. Önemli noktalardan biri her zaman Probka’dan elde ettiğimiz sonuca güvenmemeliyiz. Normalde pKa hesapları uzun ve zahmetlidir. Bu yüzde çıkan sonuçları kontrol etmeliyiz. Sonuca göre eğer elde edilen pKa değeri ortamın pH’sından düşük ise proton vermiş, eğer ortamın pH’sından -biz burada 7 olarak aldık- yüksek ise proton almış olarak düşünmeliyiz. Ayrıca elde ettiğimiz sonuçlarda çok olağan olmayan değerler elde edersek de bu amino asitlerin kontrollerinin yapılması gerek.
Probka’dan elde ettiğimiz sonuçlar aşağıda görülebilir. Burada pKmodel olarak yazan değerler, o amino asitin normal pH’da olan pKa değeridir. Ortadaki pKa ise bizim yapıdaki amino asitin pKa değeridir. Burada, GLU404’ün değeri normal değerinin yukarısında ve pH değerine yakın çıktığı için incelenmesinde yarar var. HIS’ler her zaman kontrol edilmesi gerekiyor. Çünkü histidinin protonasyonu normal pH’e çok yakın olduğu için protonasyonu çabuk değişim gösterebiliyor. Ayrıca bizim sistemde CYS440 ve CYS495 için elde edilen değerler ise normal pKa değerinden ciddi derecede farklı olduğundan incelenmesi gerek. Diğer değerler ise Probka’nın bir kaç birim hata yaptığını düşünsek bile çok bir değişiklik göstermeyeceğinden kabul edebiliriz.
SUMMARY OF THIS PREDICTION
RESIDUE pKa pKmodel RESIDUE pKa pKmodel
ASP 383 A 2.84 3.80 ASP 412 A 4.63 3.80
ASP 422 A 4.77 3.80 ASP 434 A 4.56 3.80
ASP 437 A 3.52 3.80 ASP 450 A 2.33 3.80
ASP 458 A 3.78 3.80 ASP 459 A 1.14 3.80
ASP 475 A 2.86 3.80 ASP 484 A 2.43 3.80
ASP 488 A 3.43 3.80 ASP 510 A 2.96 3.80
GLU 385 A 3.50 4.50 GLU 392 A 4.70 4.50
GLU 393 A 4.86 4.50 GLU 404 A 6.38 4.50
GLU 410 A 3.56 4.50 GLU 427 A 5.49 4.50
GLU 456 A 4.99 4.50 GLU 468 A 3.44 4.50
GLU 507 A 3.79 4.50 GLU 520 A 4.79 4.50
C- 521 A 3.50 3.20 HIS 409 A 5.15 6.50
HIS 413 A 6.64 6.50 HIS 457 A 6.16 6.50
HIS 477 A 5.02 6.50 HIS 480 A 5.72 6.50
CYS 440 A 4.13 9.00 CYS 495 A 15.62 9.00
TYR 378 A 10.81 10.00 TYR 395 A 11.82 10.00
TYR 405 A 10.14 10.00 TYR 411 A 10.33 10.00
TYR 420 A 13.06 10.00 TYR 436 A 11.59 10.00
TYR 494 A 11.71 10.00 TYR 514 A 11.53 10.00
LYS 379 A 10.39 10.50 LYS 380 A 10.23 10.50
LYS 387 A 10.70 10.50 LYS 416 A 10.51 10.50
LYS 435 A 8.23 10.50 LYS 452 A 11.22 10.50
LYS 489 A 10.69 10.50 LYS 502 A 10.64 10.50
LYS 518 A 11.35 10.50 ARG 388 A 12.44 12.50
ARG 447 A 13.07 12.50 ARG 465 A 12.62 12.50
ARG 466 A 12.30 12.50 ARG 470 A 14.41 12.50
ARG 472 A 12.33 12.50 ARG 487 A 12.36 12.50
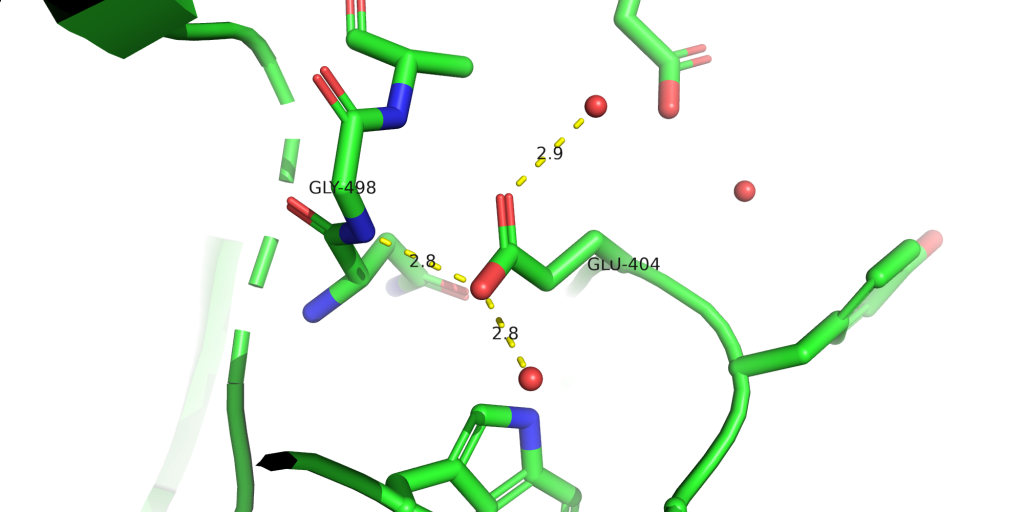
ARG 493 A 14.30 12.50 N+ 378 A 7.55 8.00Burada, GLU404 için bakacak inceleyecğiz. Eğer bir amino asit proteinin gömülü (hidrofobik) bir yerindeyse nötr olmayı isteyecektir. Ayrıca, elektrostatik etkileşimler, hidrofobik ortamda perdelenmez ya da çok az perdelenir, o yüzden yüklü olan amino asitler hidrofobik ortamda etrafındaki amino asitlerin pKa değerlerinde yakın olmasalar bile önemli bir değişime neden olur. Bu durum HIS amino asitlerinde daha çok dikkat edilmesi gerek bir durum. Burada bizim yapımızda GLU kısmen gömülü diyebileceğimiz bir bölgede olduğu için pKa’sında artışa neden olmuş olabilir. GLU’nun etrafındaki 4 Å mesafede olan amino asitlerle olan etkileşimleri incelememiz gerekiyor. Burada cevaplamamız gereken ilk soru, GLU’nun iyonize olmuş halini stabilize edebilecek amino asitler var mı ? Sonuç olarak, 3 tane hidrojen bağı yapabilecek durum söz konusu ve probkadan elde ettiğimiz sonucun makul olduğunu görüyoruz. Eğer proton alması gereken bir durum olduğunu düşünseydik ismini pdbde GLH olarak değiştirilmesi gerekecekti.
HIS için ise hepsinin kontrol edilmesi gerekiyor demiştik. Ama burada bir tanesini örnek olarak göstereceğiz. HIS için 4 farklı durum söz konusudur. İlki histidinin hem delta hem epsilon azotunda proton olmadığı hal -ki bu çok istisna bir durum olmadıkça olmuyor, o yüzden düşünülmesine gerek yok-, ikinci olarak deltada proton var epsilonda yok (ismini HID yapmalıyız), üçüncü olarak ikinci durumun tam tersi (ismini HIE yapmalıyız) ve son olarak ise ikisinde de proton var (ismini HIP yapmalıyız). HIS409 için baktığımızda, amino asitin bulunuduğu yerin kısmen su erişebileceği bir bölgede olduğunu görüyoruz. Ayrıca TYR411’in ana yapısındaki (backbone) azotu ile de bir hidrojen bağı yaptığı gözüküyor. Bir backbone azotta bir hidrojen olacağından HIS deltasında hidrojen olmamasını bekleriz. Bu yüzden epsilon azot üzerinde hidrojen var diyebiliriz ve ismini HIE olarak değiştiriyoruz. Diğer HIS’leri de bulunduğu lokasyonun solvent erişebilirliği ve etrafındaki etkileşimler açısından değerlendiriyoruz. Sonuç olarak, HIS413 için HID, HIS457 için HID ,HIS477 için HIP ve HIS480 için HIP olarak histidinleri tanımlamış olduk. Buradaki isim değiştirme işleminde önemli meselerden biri, eğer histidin HIS olarak bırakılırsa AMBER bunları otomatik olarak HIE olarak tanımlıyor. Bu yüzden istenmeyen bazı durumlar olabilir. Bu işlemleri yapmamızın en temel sebebi X-ray çalışmalarında hidrojenler görülmez, bu yüzden de bu atomların durumunun değerlendirilmesi gerekmektedir.
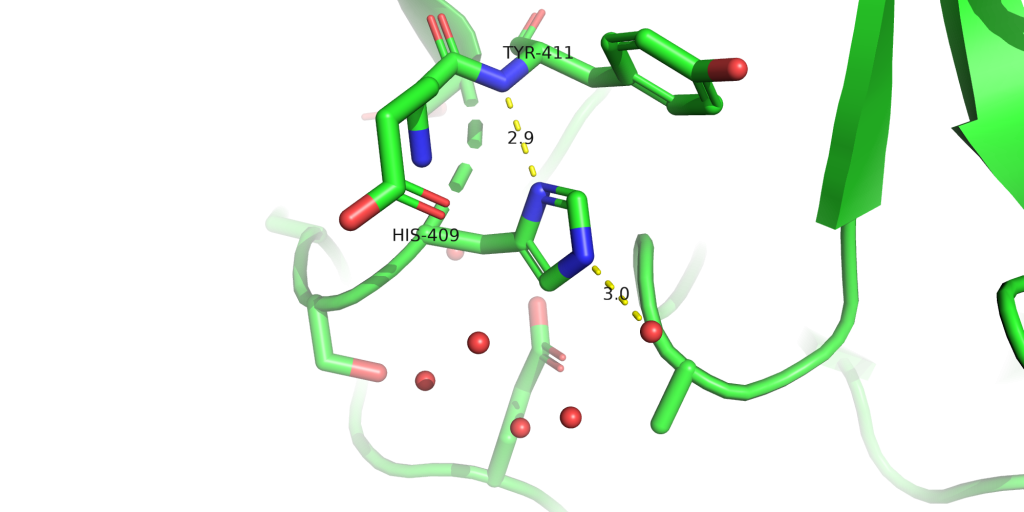
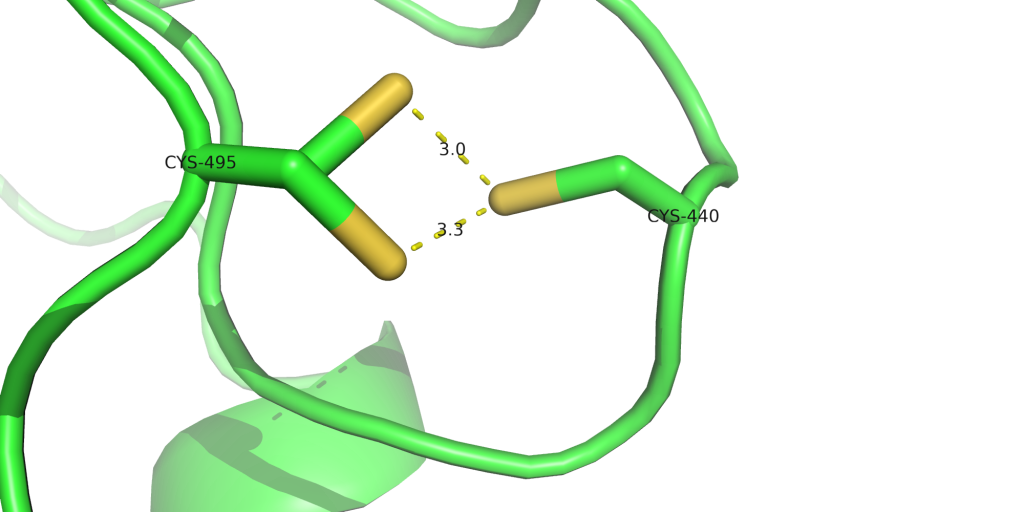
CYS440 ve CYS495 için bakacak olursak. X-ray de CYS495 için iki tane durum çözümlenmiş olarak duruyor. Bunlar, pdb içinde ACYS ve BCYS olarak tanımlanır. Bizim istediğimiz durum hangisi ise A ya da B den birinin olduğu tüm atomlar silinir ve geriye kalanların isimleri standart isimleriyle değiştirilir. Bizim durumumuzda BCYS disulfat bağ yapabilecek şekilde duruyorlar. Bu yüzden ACYS’e ait atomları sildik. BCYS isimini ise CYX olarak değiştirilmesi gerekiyor. Bundan dolayı CYS440 adını da CYX olarak değiştirilir. Eğer disulfat bağını istemeseydik BCYS’e ait atomlar silinir ve isimleri CYS olarak değiştirilirdi.
Bizim şuan incelediğimiz sistemde, pdbde yapılması gerekilen bir diğer değişiklik ise SER500 için yapılması gerek. Yukarıdaki CYS495’deki gibi burada da iki farklı durum için yapı çıkarılmış ve bunlardan birini tercih etmemiz gerek. Biz burada tamamen isteğe bağlı olarak ASER’i seçittik ve diğer atomları siliyoruz. ASER adını da SER olarak değiştiriyoruz.
- Simülasyonda Kullanılacak Dosyaların Hazılanması
Bu aşamadan sonra ise hazırladığımız pdb dosyasını kullanarak topoloji ve koordinat dosyalarını oluşturacağız. Bu dosyaları minimizasyon ve simülasyon için kullanacağız. Bunun için AMBER’de bulunan “tleap” programını kullanacağız. Aşağıda topoloji ve koordinat dosyalarının oluşturulduğu kodlar yazılı.
[csaylan@local ]$ tleap
>source leaprc.protein.ff14SB
>source leaprc.water.tip3p
>x = loadpdb 1l1d_chainA.pdb
>solvateoct x TIP3PBOX 10
>charge x
>addions K+ 0
>bond x.440.SG x.495.SG
>saveamberparms x 1l1d_chainA.top 1l1d_chainA.incrdBurada “source” ile başlayan komutlar ile, su ve protein için gereken parametreleri yüklüyoruz. Protein için günümüzde sıklıkla kullanılan ff14SB force-field’ını ve su için ise tip3p su tipinin parametrelerini ekledik. “loadpdb” ile pdb dosyamızı x adıyla tleap içinde tanıttık. “solvateoct” komutu ile trancated octahedron yapıda su ile dolu bir kutu oluşturduk. Bu kutunun boyutunu da proteinin yüzeyinden 10 Å mesafede olacak şekilde ayarladık. Kutunun nasıl alınması gerektiği çalışmadan çalışmaya önemli olabilir. Çünkü kutu içinde tamamen su ile kaplanacağı için sistemin toplam atom sayısı artacaktır. Toplam atom sayısına bağlı olarak da simülasyon için gereken zaman değişkenlik gösterecektir. Genelde küp şeklinde kutular alınır. Biz küpün kenerlarının kesilmiş olduğu yapı olan trancated octahedron kullandık, böylece bir miktar da olsa ihtiyaç olmayan suları silmiş olduk. Bu bizi simülasyondaki hesapsal iş yükünden bellirli bir oranda kurtaracaktır. Aynı zaman da ilerleyen aşamada anlatacağım periodik kutular arasında boşluk olamamasını da sağlamış olacağız. “charge” komutu bizim sistemimizin toplam yük miktarını veriyor. Bizim sistemimizin yükü -3. “addions” komutu ile net yükü sıfır yapmak için karşı iyon olarak K+ ekliyoruz. “bond” komutu ile de disulfat bağ yapacak CYS amino asitlerinin SG atomları arasında bağ olduğunu AMBER’e söylüyoruz. Son olarak ise bu yaptıklarımızı top ve incrd adında iki dosya olarak kaydediyoruz. Bu iki dosya minimizasyonda ve simülasyonda kullanılacak.
- Minimizasyon
Bu aşamadan sonra minimizasyona geçiyoruz. Minimizasyon bir tür geometri optimizasyonudur. Başlangıç yapımızda olabilecek “steric clash” denilen atomların üst üste gelme durumundan kaynaklı , ve aynı zamanda atomlar arası bağların kısalığı ya da uzunluğundan kaynaklı enerjideki yüksek dalgalanmayı önlemek için sistemin enerjisini, üzerinde bulunduğu potensiyel enerji yüzeyindeki bir minimuma getirmek gerekiyor. Bu minimum bizim sistem geometrisinin en iyi olduğu bir yer olmasa da, simülasyonun başlaması için makul bir yer olacaktır. Eğer bu işlemi yapmazsak, enerjinin yüksekliğinden kaynaklı atomlar üzerindeki kuvvet ve -haliyle- hızların çok yüksek çıkmasına neden olacak. Bunun sonucunda da sistemimiz bir süre sonra dağılıp simülasyonun başarısız olmasına neden olur. AMBER için minimizasyonu yapmak için bir “.in” uzantılı bir dosya hazırlamamız gerekiyor. Aşağıda in dosyasının içeriği ve ayrıntıları yazıldı.
minimization
&cntrl
imin=1, ntmin=1, ncyc=250, ntb=1, cut=9,
drms=0.01, maxcyc=100000
&end“in” dosyamızın içinde ilk satır başlık satırıdır. Burayı kendimiz için not bıraktığımız bir alan olarak düşenebiliriz. Burada birden fazla satırdan oluşan bir yazı da yazabilirdik. “&cntrl“ den sonra AMBER’de ne yapılmasını istediğimizi yazmaya başlıyoruz. her bir kod arasında virgül olmak zorunda ve kodların sırası önemli değil. “imin=1” ile minimizasyon yapmak istediğimizi belirtiyoruz. “ntmin=1“ ile de hangi minimizasyon algortihması kullanmak istediğimizi belirtiyoruz. Birden fazla minimizasyon algortihması var. Bunlar “non-derivative“, “first-derivative” ve “second-derivative” algoritmalar. “non-derivative” algoritmalar neredeyse hiç kullanılmaz, kullanılacaksa da elimizden hiçbir şey gelmiyor dediğimiz anlarda kullanılır, fakat kullanılması çok tavisye edilmez. Bu algoritmalara “simplex” örnek olarak verilebilir. “first-derivative” algoritmalar için “steepest descent” ve “conjuge gradient” örnek olarak verilebilir. “steepest descent” geometri minimumdan (aynı zamanda kuyunun dibi olarak da adlandırılır) uzakta ise bizi kuyunun dibine hızlıca yaklaştırır. Yalnız, hesaplama için kullandığı algoritmadan dolayı minimuma yaklaştıkça bulmakta zorlanır. “conjuge gradient” ise tam tersi kuynun dibinde uzak ise yaklaşmak uzun sürer ama minimuma yaklaşında minimumu bulmak kolaylaşır. Bu yüzden genelde önce bellirli bir adım kadar “steepest descent” yapılır ve sonra “conjuge gradient” algoritmasına geçiş yapılır. “ntmin=1″ komutu ile biz bunu AMBER’de tanımlamış oluyoruz. (Algoritmaların çalışmasını ve ayrıntılı bilgi için Andrew R Leach – Molecular modelling principles and applications (2001) kitabını inceleye bilirsiniz). “ncyc=250” kullanarak bizim kaç adım kadar “steepest descent” uygulayacağımızı söylüyoruz. Bu adımdan sonra “conjuge gradient” algortihmasına geçiş yapıyoruz. “ntb=1” ise periodik kutu kullanacağız demek oluyor. Periodik kutu, sonsuz sayıda bizim proteininimizin bulunduğu kutunun replikasını yanyana üst üste getirilmesi ile oluşan yapıya deniyor. Bunun bize avantajı ise, sonsuz bir solvent ortamı yaratmamızı sağlıyor. Ama kullanılmayadabilinir, özellikle implict ve vakumda yapılan simülasyonlarda kullanılmaz. Aşağıdaki şekil periodik kutu işleyişi gösteriyor.
Elektrostaik etkileşimler “long range” denilen uzun mesafelerde bile etkisi görünen etkileşimlerdir. Bu etkileşimleri çok uzaklardaki atomlarda bile hesaplamak fazlasyıla performans gerektirecek bir iş olacağı için Particle Mesh Ewald yöntemini kullanacağız. Bu yöntem sadece periodik kutu var ise uygulanabilir. Bu yöntem ile atomların etrafına belirli bir yarı çapta küreler çizilir. Bu atomun üzerindeki elektrostatik etki bu küre içinde kalan atomlar ile normal olarak hesaplanır. Bu alan dışındakilerin etkisi ise yaklışık olarak eklenir. Bu yarı çap 8-10 Å arasında olması yeterlidir. Eğer periodik kutu kullanılmayacak ise elektrostatik etkileşimler için gereken yarı çap 30 Å’dan düşük olmaması gerekiyor. “cut = 9” komutumuz ile de 9 Å çapında bir küre kullanacağımızı belirtmiş oluyoruz. Aynı zamanda bu komutu kullanarak van der Waals etikleşimleri de bu küre içinde normal hesaplıyoruz ama küre dışını direk sıfır olarak hesaplamak istediğimizi belirtiyoruz.

“drms=0.01“; enerjinin minimum oladuğu nokta, potensiyel enerji yüzeyindeki bir kuyunun dibidir. Bu kuyunun dibinde enerjinin birinci türevi sıfır olması gerekiyor. Fakat bizim hesaplarımızda tam olarak sıfırı bulmak uzun zaman alabileceğinden belirli bir “convergence” dediğimiz yakınsama kriterine kullanmamız gerekiyor. Bu kriterin aşağısına geldiğimizde minimizasyonu duruduruyoruz. Bu yakınsama kriteri Root Mean Square Gradient (RMSG) değeridir ve bu değere bakarak minimizasyonun ne zaman bitmesini istediğimize karar veriyoruz. Ayrıntı için Andrew R Leach – Molecular modelling principles and applications (2001) kitabını inceleye bilirsiniz). Burada 0.01’lik bir yakınsama kriteri koyduk. “maxcyc=100000” sistemimiz oldu ki hatadan kaynaklı bir tekrara girdi o zaman maksimum adım değeri belirliyoruz ki sistemimiz bir yerde durabilsin, bu komutta da bunu belirttik. Yazacağımız tüm komutlar bitince de sonuna “&end” diyip sistemi minimizasyon için çalıştırmaya başlayabiliriz.
- HPC iş gönderimi
Bu işlemler yüksek performas gerektirdiğinden ya özel hazırlamış iş istasyonlarında ya da bu işler için kullanılabilecek merkezlerdeki süper bilgisayarlarda iş gönderme usulüyle çalıştırılır. Bu merkezlerde genelde iş gönderimi Slurm İş Yükü Yöneticisi ile düzenlenir. Aşağıda minimizasyon için basit bir slurm iş gönderme betiği bulabilirsiniz. (örnek betik dosyaları için bknz. TRUBA kullanıcı el kitabı). Bu merkezlerde size tanımlı cpu time ve belli bir miktar dosya depolama alanı veriliyor. Bu alan ve zamandan harcayarak, işleri özelleşmiş kuyruklarda çalıştırıyorsunuz.
#!/bin/bash
#SBATCH -J 1l1d
#SBATCH -A csaylan # account / proje adi
#SBATCH -n 20 # cekirdek / islemci sayisi
#SBATCH -N 1 # node sayisi
#SBATCH -p mid1 # kuyruk
module load centos6.4/app/amber/14-openmpi-gcc
module load centos6.4/lib/openmpi/1.6.5-gcc
mpirun -np $SLURM_NPROCS $AMBERHOME/bin/pmemd.MPI -O -i minimization.in -c 1l1d_chainA.incrd -p 1l1d_chainA.top -o 1l1d_chainA.out -r 1l1d_chainA_min.rst
- Simülasyon
Minimizasyon başarı ile bittikten sonra, simülasyon aşamasına geçebiliriz. Simülasyon için de mizimizasyon gibi bir in dosyası hazırlamamız gerekiyor. Aşağıda komutları detaylarıyla bahsettim.
simulation
&cntrl
imin = 0, cut = 9, ntb = 2, ntp = 1,
ntt = 3, gamma_ln = 5, tempi = 5, temp0 = 310,
dt = 0.002, ntc = 2, ntf = 2, nstlim = 2500000,
ntwx = 2500, ntwe = 2500, ntpr = 100,
&end
&ewald
nfft1 = 90, nfft2 = 90, nfft3 = 90,
&end“imin = 0” komutu simülasyon yapacağımızı ifade ediyor. Burada biz NPT simülasyonu yapcağız. NPT simülasyonlarında, sistemdeki tanecik sayısının sabit ve sıcaklık ve basıncı dengelemek için sistemimize müdahale yaptığımız simülasyonlardır. NVE ve NVT gibi başka yöntemler de var. “ntb = 2” komutu sabit basınçta simülasyonu yapmamızı ifade ediyor ve bu komut 2 yapıldığında her zaman ntp > 1 olacak bir komut eklenmesi gerekiyor. Ayrıca “ntb=2” minimizasyonda kullanılmaz çünkü minimizasyonda basınç tanımlı bir kavram değil. Bu komut ile basıncı ayarlamak için kullanacağımız “isotropic scaling” olarak bilinen algoritmayı kullanacağımızı ifade ediyoruz. Burada basınç hacim ile oynayarak sabitleniyor (diğer seçenekler için için bknz. AMBER Manual). “ntt = 3” komutu sıcaklığı ayarlamak için bir termostat kullandığımızın ifadesidir. 3 ile Langevin dynamics kullandığımızı gösteriyoruz. Langevin dynamics, sisteme sanal çarpmalar yaparak sıcaklığı arttırıyor. “gamma_ln = 5” komutu ise bu sanal çarpmaların frekansını ifade ediyor. Bu değer 1/ps cinsindendir ve bu çarpmaları ne kadar sıklaştırırsak sistemin bozulmasına neden olacağından makul bir sıklık alınması önemlidir, bu yüzden 2-5 arası bir değer yeterlidir. Sistem büyüdükçe de bu değeri 2-3 civarında seçmek daha iyi olacaktır. “tempi = 5, temp0 = 310” komutlarıyla sistemin sıcaklığının 5 K’den başlayıp 310 K’e ulaşmasını ve bu sıcaklıkta devam etmesini istediğimizi söylüyoruz. “dt = 0.002” ile time step dediğimiz adım aralığını belirliyoruz. Bu değer ps cinsindendir. Bu değer yüksek sıcaklıklarda bağlar arasındaki frekans artacağından düşürülmesi gerek. Çünkü bu zaman aralığı süresince hız sabit alınır ve uzun bir zaman aralığında sistemde bozulma meydana gelir. “ntc = 2” bu komutu ile SHAKE algoritmasını kullanmak istediğimizi söylüyoruz. SHAKE algoritması ile ağır atom hidrojen arasındaki “bond stretching” hareketi sabitlenir. Eğer SHAKE kullanılmayacaksa, zaman aralığı 0.001 alınması gerekiyor. “ntf = 2” komutu ile de sabitlediğimiz bu bağların enerji hesaplarının yapılamamsını istediğimizi söylüyoruz. Çünkü hiç değişmeyeceğinden sürekli hesap yapmak iş yükünü arttıracaktır. SHAKE algoritmasında ağır atom ağır atom arasındaki bağlar için de seçenekler var ama çok tavsiye edilmez. Çünkü sistemin konformasyonel değişimine zarar verir. Ayrıca SHAKE yapılabileneceği her durumda yapılmasında fayda vardır ve sıklıkla görebilirsiniz. Fakat minimizasyon sırasında SHAKE kullanmak doğru değildir. Bunun nedeni de biz hidrojenleri sonrasından eklediğimizden bundan kaynaklı hataları önlemek. “nstlim = 2500000” bu komut sistemimiz kaç adım ilerlemesini istediğimizi bir diğer ifade ile simülasyon süremiz ne kadar olması gerek bunu söylüyoruz. Simülasyon süresi nstlim * dt şeklinde hesaplanır. Biz burada 5ns süreli bir simülasyon yapılmasını söyledik. Eğer simülasyonda çok uzun sürelere çıkacaksa bunu parçalar halinde yapılması hem sistemin karşılaşabileceği hatalardan kaynaklı veri kaybını önlemesini, hem de çok yüksek boyutta yer kaplayan bir dosyayı parçalara ayırarak taşımasını ve saklanabilirliğini arttırmayı sağlayacaktır. “ntwx = 2500, ntwe = 2500, ntpr = 100” bu komutlar ile belirli adımlarda sistemimizin koordinat ve sistem enerjisi gibi işe yarar bilgilerini yazılmasını söylüyoruz. ntwx koordinatlar için, ntwe enerji verileri için, ntpr daha anlaşılabilinir bir dil ile sistemin özetini içeren bir out dosyası çıkarması için kullanılır. Bu çıktılar sistem içi yanlışlıklarını anlayabilmemiz için önemlidir, bu nedenle özellikle ntpr için aldığımız aralığın daha az alınmasında fayda vardır. Burada çok sık yazım yapılırsa elimizde çok yüksek boyutlarda veri olabileceğinden seyrek aralıklarla ama Bu adım aralıkları arasındaki tüm veri kaybedilmiş olacak o yüzden de çok seyrek aralıklar seçilmemeli. Sonrasında da “&end” komutları bitiriyoruz. Ekstra komut için yeni bölüm tanımlamamız gerekiyor. Simülasyon in dosyamızda, PME için &ewald diyerek yeni bir bölüm açıyoruz. Burada belirttiğimiz komutlar “nfft1 = 90, nfft2 = 90, nfft3 = 90“. Bu komutları PME’de kullanılacak olan “charge grid”lerinin boyut büyüklüklerinin ifadesi için kullanıyoruz. Bu değerleri AMBER otomatik hesaplıyor. Bu değerleri minimizasyon çıktısında aratarak bulabilirsiniz. Daha detaylı bilgi için bknz. AMBER Manual. Bu değeri yüksek almak PME’nin doğruluğunu arttırırken, simülasyonun işlem yükünü arttırıcağından yavaşlatır. Tam tersinde ise simülasyon hızı artarken, PME’nin doğruluğu azalacaktır. Bundan sonra &end diyip bu bölümü kapatıyoruz. Sisteme restraint gibi ifadeler eklemek için de bu şekilde bölmeler açılıp gerekli olan komutları eklememiz gerekiyor.
Bu aşamadan sonra sistem, simülasyon yapılması için hazır durumda. Sistemi çalıştırmak için minimizasyonda yaptığımız gibi bir slurm betiği hazırlamak veya kendi iş istasyonumuzdan işi çalıştırabiliriz. Örnek olarak simülasyon betiği aşağıda bulabilirsiniz.
#!/bin/bash
#SBATCH -J 1l1d
#SBATCH -A csaylan # account / proje adi
#SBATCH -n 20 # cekirdek / islemci sayisi
#SBATCH -N 1 # node sayisi
#SBATCH -p mid1 # kuyruk
module load centos6.4/app/amber/14-openmpi-gcc
module load centos6.4/lib/openmpi/1.6.5-gcc
mpirun -np $SLURM_NPROCS $AMBERHOME/bin/pmemd.MPI -O -i simulation.in -c 1l1d_chainA_min.rst -p 1l1d_chainA.top -o 1l1d_chainA_1.out -r 1l1d_chainA_1.rst
Eğer sistemi gpu kullarak slurm üzerinden çalıştıracaksanız module load centos6.4/lib/cuda/5.5 gibi cuda için ilgili module yüklenmeli ve #SBATCH –gres=gpu:1 eklenerek sistemin kaç gpu kulanmasını istediğimizi belirtmeliyiz. Bunlara ek olarak pmemd.cuda komutu kullanılmalıdır.
Sistem çalışmasını bitirince de kordinat dosyalarını cpptraj gibi analiz yapabileceğiniz programlarla analizini yapabilirsiniz.
Referanslar
- Hollingsworth, S. A., & Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron, 99(6), 1129-1143.
- Computational Biology: Structure and Organization of Biomolecules and Cells | Stanford Online. (2020). Alıntı tarihi 15 Temmuz 2020, http://web.stanford.edu/class/cs279/
- Leach, A. R., & Leach, A. R. (2001). Molecular modelling: principles and applications. Pearson education.